EGFL7 meets miRNA-126: an angiogenesis alliance
Journal of Angiogenesis Research. 2010;
Received: 21 April 2010 | Accepted: 8 June 2010 | Published: 8 June 2010
Vascular Cell ISSN: 2045-824X
Abstract
Blood vessels form
Introduction
Vasculogenesis and angiogenesis are basic processes through which new blood vessels arise. Vasculogenesis entails the differentiation of mesodermal cells into endothelial precursor cells (angioblasts). This is followed by the formation of primitive blood vessels which are subsequently refined and transformed into a functional vascular network by the process of angiogenesis [1, 2]. The execution of these tightly regulated programs depends on a vast array of factors whose identification has been a prime focus of cardiovascular research in the last two decades. One of the newly described molecular players in the field of blood vessel formation is EGFL7, a protein also known as VE-statin, MEGF7, Notch4-like protein or Zneu1. Initial studies on the role of EGFL7 in the vascular system were compiled by Soncin et al., who showed that EGFL7 inhibited the migration but not the proliferation of human aortic smooth muscle cells
EGFL7 structure, temporal and spatial distribution
Initially, EGFL7 was described as a 30 kD protein exclusively expressed by vascular endothelial cells [3]. The protein is conserved among vertebrates but an orthologue is also found in
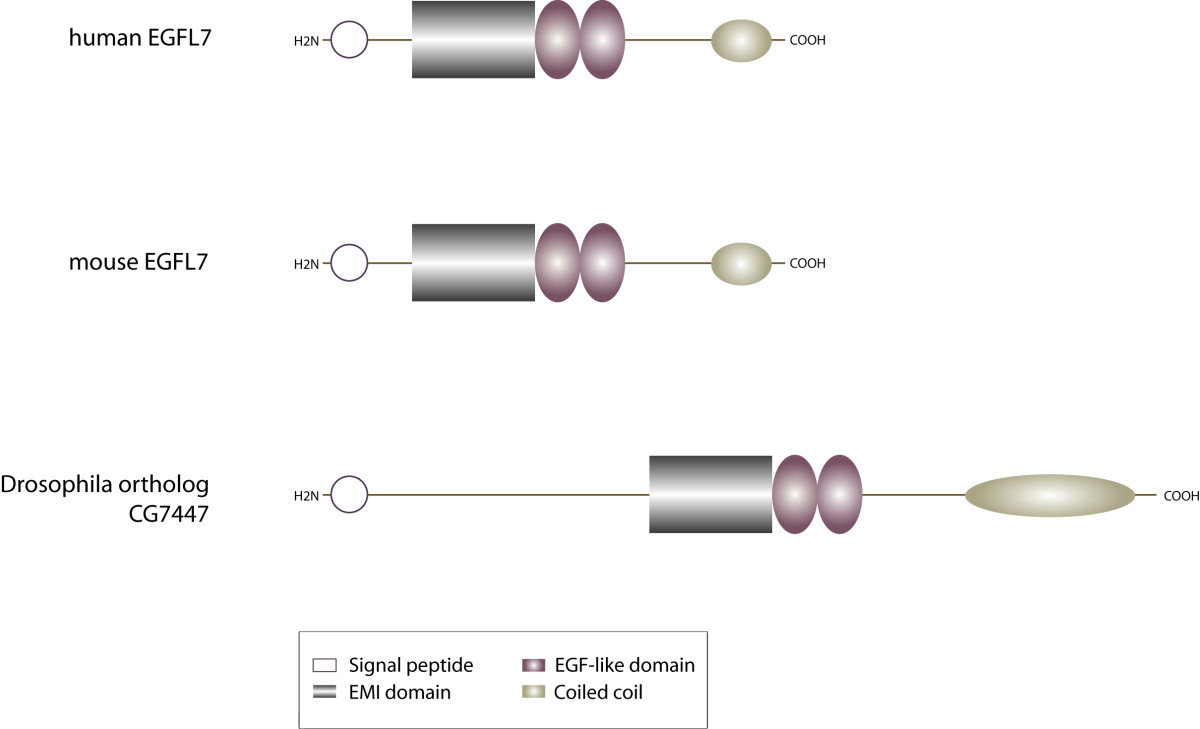
Figure 1
Figure 1 caption
Domain organization of EGFL7. The modular assembly of EGFL7's domains is conserved among vertebrate species. The protein contains an N-terminal signal secretion peptide, an EMI domain, two EGF-like domains (the distal one binds Ca) and a C-terminal coiled-coil structure. The EGFL7 orthologue CG7447 in shares the same overall structure but differs in total length.
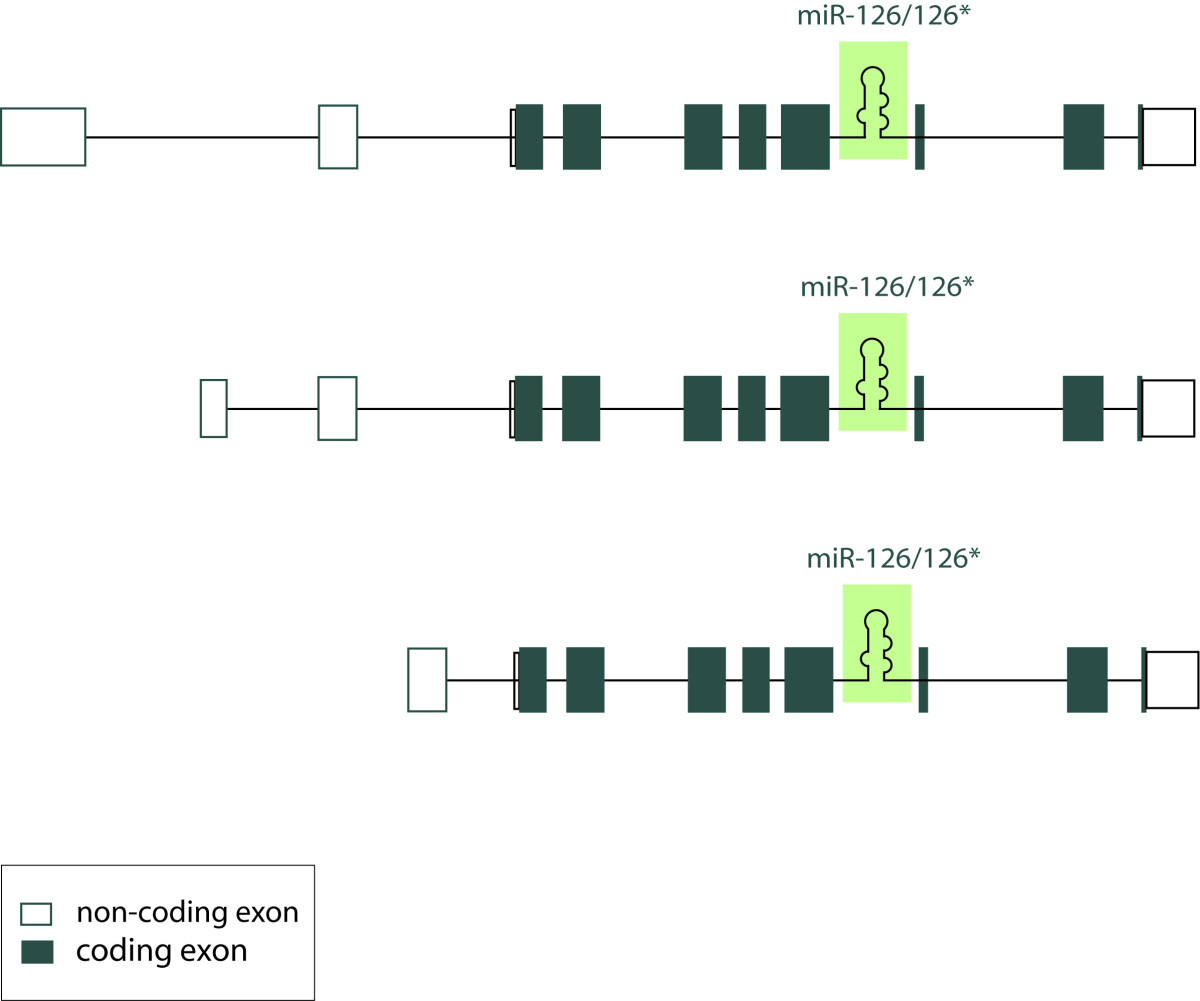
Figure 2
Figure 2 caption
Localization of miR-126/miR-126* within the primary human EGFL7 transcript. This is a schematic representation of three alternative primary human EGFL7 transcripts, which initiate from separate promoters but contain the same open reading frame. Non-coding exons are indicated in uncolored boxes, coding exons in colored ones. miR-126 and miR-126* are localized within intron 7 of EGFL7 in all vertebrates.
In vertebrates the
Initial expression analyses indicated EGFL7 is restricted to the vascular endothelium at all stages of mouse development [5, 13]. However, later work by Campagnolo et al. demonstrated the presence of EGFL7 in the primordial germ cells during homing into the gonads [14]. Most importantly, EGFL7 is expressed within the neurons of adult mice, which suggests EGFL7 serves diverse biological functions in various tissues and not only in the vascular system [15]. EGFL7 becomes detectable at the blastocyst stage during mouse development with a marked increase in expression levels at embryonic day E7.5 to E8.5. Subsequently, expression remains at a constant level [3, 5]. Upon birth, EGFL7 becomes downregulated in the vascular system and significant levels of the protein are only maintained in a subset of vessels in the lung, heart, kidney, spleen and uterus. High expression levels of EGFL7 are regained upon the onset of physiological angiogenesis, e.g. in the uterus during pregnancy [13] or alternatively, under pathological conditions of vessel formation subsequent to arterial injury [13], hypoxic insult [16] or in human solid tumors [4]. In sum, the striking temporal and spacial expression pattern of EGFL7 indicates a function of this protein in blood vessel formation and remodeling.
EGFL7 in the extracellular matrix
The ECM is one of the key components of the vascular system as it maintains the organization and regulation of endothelial cells [17]. The ECM supports endothelial cell proliferation, migration, survival and morphogenesis during blood vessel formation. Primarily, this occurs through adhesive interactions with integrins on the endothelial cell surface. In addition, ECM proteins may function to sequester angiogenic cytokines allowing for the coordination of signals transduced via growth factor receptors and integrins [18, 19]. Several parameters indicate EGFL7 is associated with the ECM. First, when overexpressed in fibroblasts, EGFL7 is mainly detected in the ECM fraction and the cell lysates and only little amounts of the protein are spotted in the conditioned medium. This suggests EGFL7 remains largely attached to the cell surface, most likely through interactions with ECM molecules [20]. Furthermore, the deposition of EGFL7 in the ECM is facilitated by certain types of matrix proteins, such as fibronectin and collagen type I, whereas laminin and collagen type IV do not exert such an effect [20]. Last, EGFL7 has been shown to promote endothelial cell adhesion and focal complex formation although not as efficiently as classical ECM molecules like collagen or fibronectin do [4]. In a recent study, Lelievre et al. used a transgenic mouse model to express EGFL7 in the epidermis and detected a colocalization of EGFL7 and elastic fibers in the ECM of epidermal blood vessels. Such they identified EGFL7 as a negative regulator of vascular elastogenesis [21]. Elastic fibers are the largest structures in the ECM with a very complex organization. Elastin as one of the key fiber components is produced in a process that includes cross-linking of tropoelastin molecules by a family of lysyl oxidases (LOXs) [22]. The colocalization of EGFL7 with the elastic fibers caused an inhibition of the enzymatic activity of LOXL2 by direct interaction and interfered with the process of elastin deposition [21]. Enzymes of the LOX family cross-link elastin and collagen [23] therefore one may speculate that EGFL7 participates in the shaping of the ECM thereby indirectly affecting endothelial cell functions such as migration. Particularly, these findings are interesting because EGFL7 molecules harbor conserved domains that are typically associated with ECM proteins. The EMI domain is a cysteine-rich repetitive element often detected in extracellular proteins that form multimers, e.g. emilin-1 and emilin-2. Likewise, EGF-like domains are frequently found in extracellular proteins like the constituents of elastic fibers [11, 24] or common ECM molecules like laminin and tenascin [12]. Taken together, data suggest EGFL7 as a putative novel component of the ECM giving clues as to how it affects endothelial cells.
EGFL7 in vasculogenesis
Early in embryonic development several inductive cues, e.g. members of the fibroblast growth factor (FGF) or the bone morphogenetic protein (BMP) families, initiate the differentiation of hemangioblasts from the undifferentiated mesoderm [25, 26]. Hemangioblasts form aggregates in which the inner cells develop into hematopoietic precursors and the outer population eventually gives rise to endothelial cells. Subsequently, endothelial precursor cells or angioblasts differentiate and assemble into a primitive vascular network. Commonly, this process is referred to as vasculogenesis [27]. Solid evidence for a role of
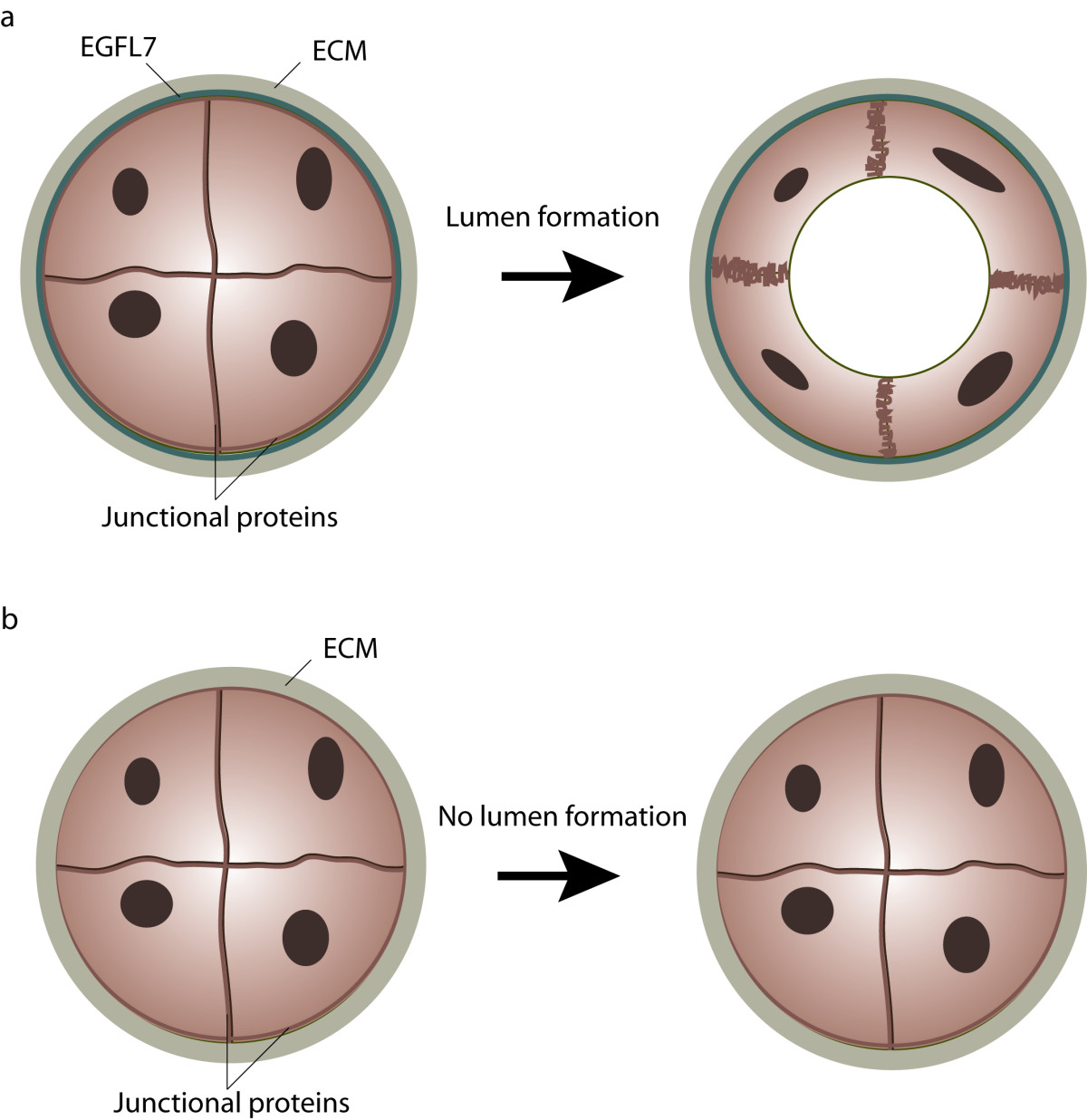
Figure 3
Figure 3 caption
Role of EGFL7 in lumen formation. (a) During primitive plexus formation angioblasts assemble into vascular cords which are subsequently transformed into vascular tubes. Cord-to-tube transition is characterized by cell polarization, redistribution of junctional proteins and eventually, changes in cell shape. EGFL7 is secreted by endothelial cells into the extracellular matrix, where it affects the process of lumen formation. (b) In the absence of EGFL7 angioblasts fail to separate, which leads to the formation of vessels lacking a vascular lumen.
EGFL7 in angiogenesis
In contrast to vasculogenesis, the process of angiogenesis involves the formation of new blood vessels from the pre-existing vascular network [33]. In most experiments, angiogenic sprouting is studied although angiogenesis also proceeds through intussusceptions [34, 35]. Angiogenic sprouting involves a number of tightly regulated steps: vasodilatation and endothelial permeability, endothelial cell proliferation and migration, lumen formation as well as endothelial cell survival and vessel maintenance [36]. Due to the role of EGFL7 in tubulogenesis [4] it is reasonable to assume a comparable role for EGFL7 in angiogenic sprouting. In order to investigate this matter, Schmidt et al. created two independent mouse lines harboring EGFL7 deletions [20]. The first line was generated by application of a retroviral gene trap vector which was inserted in intron 2, upstream of the translation initiation codon in exon 3, whereas the second mouse line was created by homologous recombination resulting in the removal of the DNA region lasting from exon 5 to exon 7. 50% of the corresponding EGFL7-/- embryos died
In order to unravel the cellular basis for the observed defects in murine vessel growth, Schmidt et al. performed a detailed analysis of angiogenic sprout morphology [20]. Namely, they studied angiogenic sprouting, which proceeds through the coordinated actions of two cell types: tip cells and stalk cells. Tip cells sense a VEGF gradient in the surrounding environment and extend filopodia, which leads the angiogenic sprout in a specific direction. The trailing stalk cells on the other hand proliferate and support sprout elongation [37]. Eventually, endothelial cells reacquire a quiescent phenotype, recently referred to as phalanx cells, which mediate the stabilization of newly formed vessel [38].
In wild type animals tip and stalk cells organize within a single cell layer, while in EGFL7 knock-out mice both cell types form multiple cell layers similar to the cell aggregates observed in EGFL7 knock-down zebrafish embryos [4]. Immunohistochemical staining of collagen IV revealed that this ECM molecule, which is typically localized in the basal membrane, was found within these enlarged sprouts and was detected between the adjacent endothelial cells. This suggests that endothelial cells lacking EGFL7 failed to properly detect the sprout boundaries. Previously, it has been shown that EGFL7 supports the weak adhesion of endothelial cells [4], suggesting EGFL7 creates an environment where cells easily attach and detach until properly positioned. In the absence of EGFL7, however, cells may clump together and build oversized sprouts resulting in an impaired migration and delayed vascularization as observed in EGFL7-/- mice.
A recent study of Schmidt et al. provided another compelling clue for resolving the function of EGFL7 protein in angiogenesis [15]. Most interestingly, a link between EGFL7 and Notch signaling has been unraveled. The Notch pathway is evolutionarily conserved and governs fundamental processes such as development, cell-fate determination and differentiation [39]. Notch-mediated signal transduction is based on several key molecules: four Notch receptor isoforms (Notch1-4) and five canonical ligands of the Delta (Dll1, 3 and 4) or the Jagged type (Jagged1 and 2) [40]. The role of Dll3 has been controversially discussed and it is not considered a
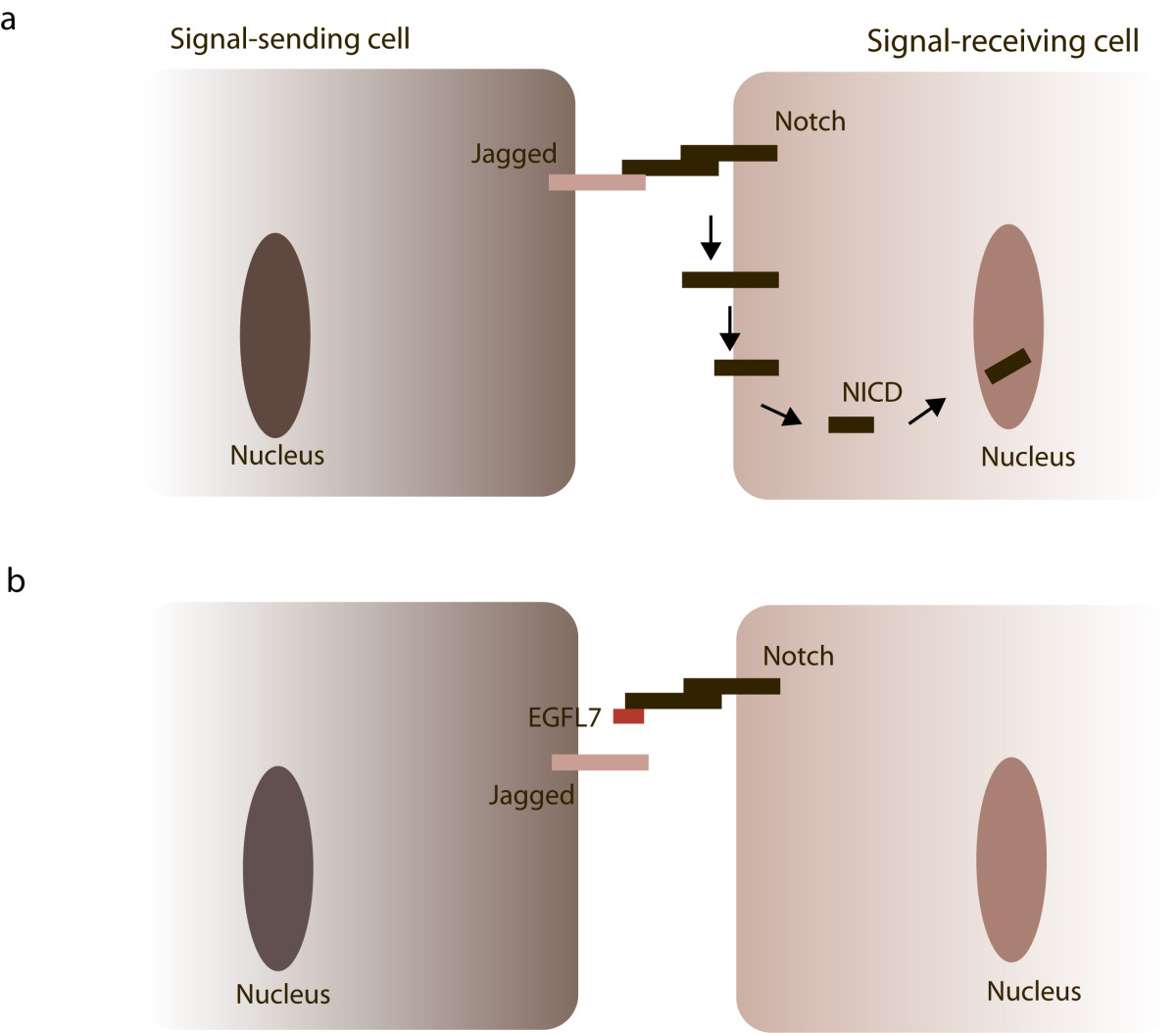
Figure 4
Figure 4 caption
EGFL7 and Notch signaling. Notch receptors are expressed on the cellular surface as heterodimers connected by non-covalent interactions. (a) Upon binding of Jagged-type ligands expressed on neighboring cells , Notch receptors undergo a series of proteolytic cleavage events releasing the NICDs of the receptors and allowing their translocation into the nucleus. (b) If EGFL7 is present in the surrounding of the cells, it competes with Jagged for Notch binding thereby attenuating Notch signaling.
miR-126 in angiogenesis
Recently, findings on the role of EGFL7 in angiogenesis have been supplemented by several papers describing a role of miR-126 in the vascular system [6, 50, 51]. Collectively, miRNAs represent a class of about 22 nucleotide long, non-coding RNAs that have been recognized in recent years as important regulators of gene expression [52]. Predominantly, miRNAs repress protein expression by the inhibition of protein translation and to a lesser extent by mRNA degradation [53]. Mounting evidence indicates the importance of miRNAs in blood vessel formation by the regulation of endothelial and smooth muscle cell functions [54, 55]. Most interestingly, miR-126 is located in intron 7 of
Two papers were published back-to-back studying the function of miR-126 in vessel development by loss of function experiments either in zebrafish [6] or mouse [50]. In both cases loss of miR-126 caused similar phenotypes
Interestingly, a recent work demonstrated that EGFL7 is a direct target of the miR-126 in lung cancer cells and hinted that this could be at least a part of an explanation for the observed effect of miR-126 on tumourogenesis [8]. Likewise, Fish et al. described a transcriptional regulation of EGFL7 in human endothelial cells by miR-126 [6]. Taken together, above findings unambiguously demonstrate that miR-126 and EGFL7 share not only a structural but also a tight functional connection in different cellular contexts.
Concluding remarks and future perspectives
Current data emphasizes a highly significant role of
Nevertheless, at the current stage there are many unresolved issues such as an imminent lack of a defined role of the EGFL7 protein in angiogenesis or an explanation of the mechanisms underlying EGFL7's effects on endothelial cells. Further, the functional relationship between the EGFL7 protein and miR-126 is not clear and one wonders if both act in synergy or antagonism. The undisputed role for EGFL7 in blood vessel formation beyond the function of miR-126 has yet to be proven and the underlying mechanism unraveled. Mice harboring a specific EGFL7 protein deletion without the confounding effect of the loss of miR-126 [51] do not show any abnormalities during angiogenic sprouting. This could potentially be explained by the upregulation of the EGFL7 homolog EGFL8 as indicated above. In this case it might prove useful to study EGFL7/EGFL8 double knock-out mice in order to understand the effects of the individual proteins on blood vessel formation. In addition, transgenic mice expressing EGFL7 under inducible conditions will help to shed light on the role of EGFL7 in angiogenesis.
Yet another interesting question to be addressed is the identification of novel EGFL7-interacting molecules on the surface of vascular cells. Given the fact that EGFL7 incorporates into the ECM, it seems possible that integrins mediate EGFL7's effects on endothelial cells, because integrins represent a class of receptors responsible for the interaction of virtually all types of adherent cells with the ECM. Integrins are indispensible for essential processes in the vascular system such as proliferation, migration or survival [61], plus, there is considerable data that suggest integrins to play a key role in the stimulation of tubular morphogenesis and the activation of endothelial cells [62, 63]. This makes them prime candidates to be studied as mediators of EGFL7's effects on endothelial cells.
Further, it seems worthwhile to study the role of EGFL7 in cancer formation and progression as the role of
In sum,
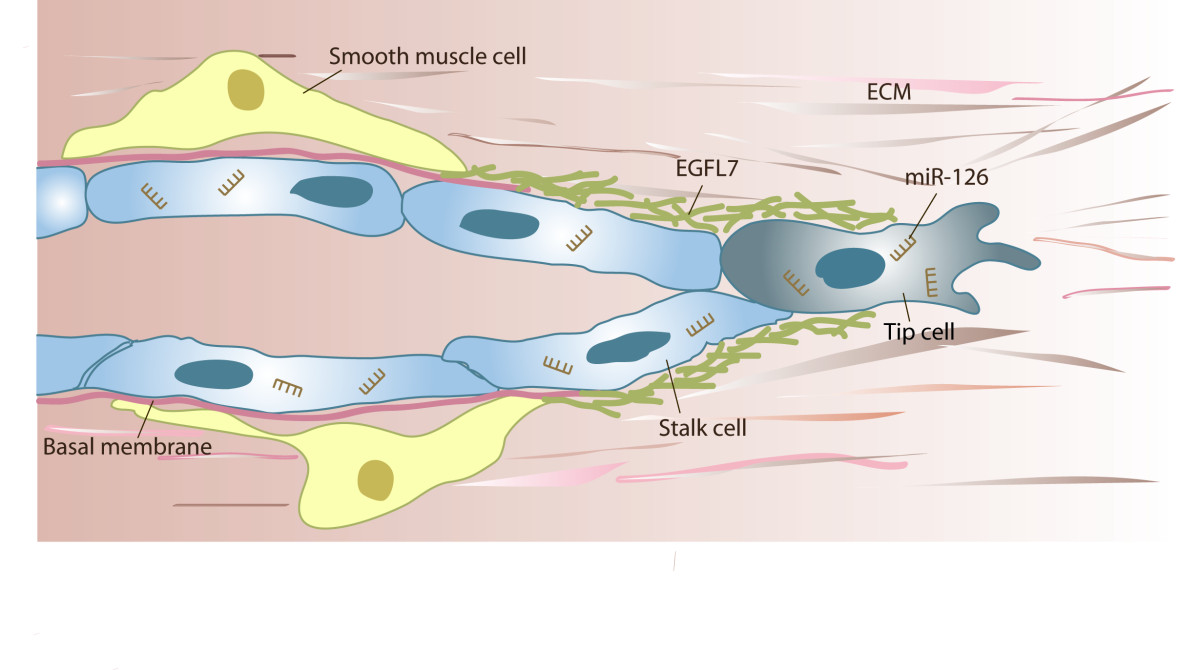
Figure 5
Figure 5 caption
Putative function ofin angiogenic sprouting. Angiogenic sprouting proceeds through the coordinated actions of tip and stalk cells. Tip cells lead the sprout through the environment, while stalk cells proliferate and thereby contribute to sprout extension. Concurrently or subsequently to invasion, sprouts lumenize, assemble a basal membrane and eventually, recruit smooth muscle cells. EGFL7 is found incorporated into the provisional ECM surrounding the sprout, whereas miR-126 targets numerous mRNA molecules within the cell, which affects proliferation and the process of angiogenic sprouting.
Acknowledgements
This work was financially supported by the German Research Foundation DFG within the framework of Transregional Collaborative Research Centre 23 (subproject A4) and the Excellence Cluster 147 "Cardio-Pulmonary Systems".
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2
Authors’ original file for figure 3
Authors’ original file for figure 4
Authors’ original file for figure 5
References
- Mechanisms of angiogenesis. Nature. 1997;386:671-674.
- Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389-395.
- VE-statin, an endothelial repressor of smooth muscle cell migration. EMBO J. 2003;22:5700-5711.
- The endothelial-cell-derived secreted factor Egfl7 regulates vascular tube formation. Nature. 2004;428:754-758.
- Egfl7, a novel epidermal growth factor-domain gene expressed in endothelial cells. Dev Dyn. 2004;230:316-324.
- miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272-284.
- Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med. 2009;13:1577-1585.
- miR-126 inhibits non-small cell lung cancer cells proliferation by targeting EGFL7. Biochem Biophys Res Commun. 2009;391:1483-1489.
- Epigenetic therapy upregulates the tumor suppressor microRNA-126 and its host gene EGFL7 in human cancer cells. Biochem Biophys Res Commun. 2009;379:726-731.
- Ectopic expression of miR-126*, an intronic product of the vascular endothelial EGF-like 7 gene, regulates prostein translation and invasiveness of prostate cancer LNCaP cells. J Mol Med. 2008;86:313-322.
- EMI, a novel cysteine-rich domain of EMILINs and other extracellular proteins, interacts with the gC1q domains and participates in multimerization. FEBS Lett. 2000;484:164-168.
- EGF-like domains in extracellular matrix proteins: localized signals for growth and differentiation?. FEBS Lett. 1989;251:1-7.
- EGFL7 is a chemoattractant for endothelial cells and is up-regulated in angiogenesis and arterial injury. Am J Pathol. 2005;167:275-284.
- Expression of EGFL7 in primordial germ cells and in adult ovaries and testes. Gene Expr Patterns. 2008;8:389-396.
- Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat Cell Biol. 2009;11:873-880.
- Vascular response to hypoxic preconditioning in the immature brain. J Cereb Blood Flow Metab. 2007;27:928-938.
- Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093-1107.
- Chen TT, Luque A, Lee S, Anderson SM, Segura T, Iruela-Arispe ML: Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J Cell Biol. 188: 595-609. 10.1083/jcb.200906044.
- Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ Res. 2002;91:25-31.
- EGFL7 regulates the collective migration of endothelial cells by restricting their spatial distribution. Development. 2007;134:2913-2923.
- VE-statin/egfl7 regulates vascular elastogenesis by interacting with lysyl oxidases. EMBO J. 2008;27:1658-1670.
- Elastic fibres. J Cell Sci. 2002;115:2817-2828.
- New insights into elastic fiber assembly. Birth Defects Res C Embryo Today. 2007;81:229-240.
- The EMILIN protein family. Matrix Biol. 2000;19:289-301.
- Induction of vasculogenesis and hematopoiesis in vitro. Development. 1992;116:435-439.
- The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci USA. 1998;95:3667-3672.
- Mechanisms of endothelial differentiation in embryonic vasculogenesis. Arterioscler Thromb Vasc Biol. 2005;25:2246-2254.
- Organogenesis: molecular mechanisms of tubulogenesis. Nat Rev Genet. 2002;3:513-523.
- The molecular basis of vascular lumen formation in the developing mouse aorta. Dev Cell. 2009;17:505-515.
- Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537-540.
- Zovein AC, Luque A, Turlo KA, Hofmann JJ, Yee KM, Becker MS, Fassler R, Mellman I, Lane TF, Iruela-Arispe ML: Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev Cell. 18: 39-51. 10.1016/j.devcel.2009.12.006.
- Durrans A, Stuhlmann H: A role for Egfl7 during endothelial organization in the embryoid body model system. J Angiogenes Res. 2: 4-10.1186/2040-2384-2-4.
- Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J Neurooncol. 2000;50:1-15.
- Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49:507-521.
- Intussusceptive angiogenesis: its role in embryonic vascular network formation. Circ Res. 2000;86:286-292.
- Angiogenesis in cancer and other diseases. Nature. 2000;407:249-257.
- VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. 2003;161:1163-1177.
- Branching morphogenesis and antiangiogenesis candidates: tip cells lead the way. Nat Rev Clin Oncol. 2009;6:315-326.
- Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678-689.
- Notch: Implications of endogenous inhibitors for therapy. Bioessays. 2010;32:481-487.
- The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. J Cell Biol. 2005;170:983-992.
- The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216-233.
- EGFL7: A new player in homeostasis of the nervous system. Cell Cycle. 2010.
- Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776-780.
- Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA. 2007;104:3219-3224.
- Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development. 2007;134:839-844.
- The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA. 2007;104:3225-3230.
- The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124-1135.
- Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell. 2005;16:927-942.
- The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261-271.
- Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development. 2008;135:3989-3993.
- MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281-297.
- Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642-655.
- MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710-1713.
- miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705-710.
- A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401-1414.
- Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59-68.
- The noncoding RNA, miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer. 2008;47:939-946.
- Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451:147-152.
- Zhong M, Ma X, Sun C, Chen L: MicroRNAs reduce tumor growth and contribute to enhance cytotoxicity induced by gefitinib in non-small cell lung cancer. Chem Biol Interact. 184: 431-438. 10.1016/j.cbi.2010.01.025.
- Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604-617.
- An integrin and Rho GTPase-dependent pinocytic vacuole mechanism controls capillary lumen formation in collagen and fibrin matrices. Microcirculation. 2003;10:27-44.
- Matrix-specific activation of Src and Rho initiates capillary morphogenesis of endothelial cells. FASEB J. 2004;18:457-468.
- Novel role for epidermal growth factor-like domain 7 in metastasis of human hepatocellular carcinoma. Hepatology. 2009;50:1839-1850.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.