Clinical trials with anti-angiogenic agents in hematological malignancies
Journal of Angiogenesis Research. 2010;
Received: 20 April 2010 | Accepted: 22 June 2010 | Published: 22 June 2010
Vascular Cell ISSN: 2045-824X
Abstract
New blood vessel formation (angiogenesis) is not only essential for the growth of solid tumors but there is also emerging evidence that progression of hematological malignancies like multiple myeloma, acute leukemias, and myeloproliferative neoplasms, also depends on new blood vessel formation. Anti-angiogenic strategies have become an important therapeutic modality for solid tumors. Several anti-angiogenic agents targeting angiogenesis-related pathways like monoclonal antibodies, receptor tyrosine kinase inhibitors, immunomodulatory drugs, and proteasome inhibitors have been entered clinical trials or have been already approved for the treatment of hematological malignancies as well and in some instances these pathways have emerged as promising therapeutic targets. This review summarizes recent advances in the basic understanding of the role of angiogenesis in hematological malignancies and clinical trials with novel therapeutic approaches targeting angiogenesis.
Introduction
The hypothesis of tumor angiogenesis in malignancies was raised by Judah Folkman: To grow over a certain size of a few millimetres in diameter solid tumors need blood supply from surrounding vessel [1]. Up to 2-3 mm3 solid tumors can grow without blood vessel supply. Nutrition and oxygen is provided via diffusion from the surrounding tissue. Above this size, diffusion becomes insufficient due to the negative surface/volume ratio. Based on a balance between angiogenic and anti-angiogenic growth factors, a tumor of this size can stay dormant for a very long time period until the so-called angiogenic switch occurs [2]. Tumor blood vessels are generated by various mechanisms, such as expansion of the host vascular network by budding of endothelial sprouts (sprouting angiogenesis), cooption of the existing vascular network, remodeling and expansion of vessels by the insertion of interstitial tissue columns into the lumen of preexisting vessels (intussusceptive angiogenesis) and homing of endothelial cell precursors (EPC; CEP) from the bone marrow or peripheral blood into the endothelial lining of neovessels (vasculogenesis) [3].
Tight control of angiogenesis is maintained by a balance of endogenous anti-angiogenic and pro-angiogenic factors [4]. VEGF has a key, rate-limiting role in promoting tumor angiogenesis and exerts its effects by binding to one of three tyrosine kinase receptors: VEGF receptor-1 (VEGFR-1; fms-like tyrosine kinase-1, Flt-1), VEGFR-2 (human kinase domain region, KDR/murine fetal liver kinase-1, Flk-1) and VEGFR-3 (Flt-4). VEGFR-1 (ligands include VEGF-A, -B and placental growth factor [PIGF]) and VEGFR-2 (ligands include VEGF-A, -C and -D) are predominantly expressed on vascular endothelial cells, and activation of VEGFR-2 appears to be both, necessary and sufficient, to mediate VEGF-dependent angiogenesis and induction of vascular permeability [4, 5]. Both receptor tyrosine kinases are expressed in all adult endothelial cells, except for the brain endothelial cells. VEGFR-1 is also expressed on hematopoietic stem cells, vascular smooth muscle cells, monocytes, and leukemic cells [6, 7], while VEGFR-2 is expressed on endothelial progenitor cells and megakaryocytes [8, 9]. VEGFR-3, largely restricted to lymphatic endothelial cells, binds the VEGF homologues VEGF-C and VEGF-D and may play an important role in the regulation of lymphangiogenesis. Thus, VEGF and VEGFR represent significant anti-cancer therapy targets, which elegantly bypass potential tumor-related treatment barriers [4].
A further important pathway in angiogenesis is the recently identified Delta-Notch pathway, and particularly the ligand Delta-like 4 (Dll4), was identified as a new target in tumor angiogenesis [10]. Dll4 is highly expressed by vascular endothelial cells and induced by VEGF [11]. It interacts with Notch cell surface receptors to act as a negative feedback inhibitor downstream of VEGF signaling to restrain the sprouting and branching of new blood vessels [10, 12]. Inhibition of Dll4-Notch signaling induces an increase in vessel density but these blood vessels are abnormal and not perfused [13]. Therefore intratumour hypoxia is increased and leads to induction of transcription of proangiogenic genes regulated by Hypoxia inducible factor-1 (HIF-1) [10, 14]. Disruption of Dll4 signaling by overexpression or inhibition of Dll4 may impair angiogenesis and blockade of Dll4-Notch signaling results in an increased density of nonfunctional vasculature and is associated with a reduction in the growth of human tumor xenografts [13, 14]. Further, certain xenografts that are resistant to anti-VEGF therapy are reported to be sensitive to anti-Dll4 and combination treatment with anti-VEGF and anti-Dll4 has additive inhibitory effects on tumor growth [13–15].
This review summarizes the role of pathological angiogenesis in hematological malignancies focusing on multiple myelomas (MM), acute leukemias, and myeloproliferative neoplasms (MPN) and its therapeutic intervention with novel agents within clinical trials or already approved.
Pathophysiology of angiogenesis in hematological malignancies
Many studies suggest a role for angiogenesis not only in the pathogenesis of solid tumors but also in hematological malignancies like acute and chronic leukemia, lymphoma, myelodysplastic syndromes, myeloproliferative neoplasms, and multiple myeloma [16–21]. We and others reported an increased microvessel density and VEGF expression in the bone marrow of patients with myeloproliferative neoplasms and lymphoma [17, 20]. Thereby, the extent of angiogenesis in the bone marrow often correlated with disease burden, progonosis, and treatment outcome [22, 23]. In the neoplastic bone marrow there is an imbalance of the cells, cytokines and growth factors maintaining physiological angiogenesis in the normal bone marrow. The bone marrow tumor cells upregulates several factors, including interleukin-6, granulocyte-macrophage colony-stimulating factor and VEGF, have autocrine and paracrine effects acting on multiple cell types, thereby stimulating angiogenesis and leading to increased vascularity [7, 24]. The role for VEGF in hematogical malignancies has been extensively studied since its isolation from the leukemia cell line HL- 60 in 1989 [25]. Apparently, this growth factor is expressed in many other leukemic cell lines [7, 26] and a subset of leukemic cells also expresses VEGFR-2 which allows VEGF to act as autocrine growth factor in leukemia [26, 27]. In addition to that, isolated blast cells from leukemia patients also produce VEGF [26] and the cellular level of VEGF in acute myeloid leukemia (AML) patients has been identified as independent prognostic risk factor [28]. VEGF from leukemic blasts contributes to disease progression, either as positive regulator for proliferation and apoptosis protection for the blast itself or by activating the surrounding stroma cells with subsequent induction of bone marrow angiogenesis.
Regarding the Notch pathway, Notch signals are oncogenic in hematogical malignancies in many cellular contexts [29]. Activating Notch-mutations have been shown to be present in at least 50% of human T-cell acute lymphoblastic leukaemia (T-ALL) cases and have been proved to play a unifying role in the pathogenesis of T-ALL [30]. An important role of Notch has been proposed in cell survival in several B-cell malignancies such as Hodgkin's disease [31, 32] and in two B-cell non-Hodgkin lymphoma entities, chronic lymphocytic leukaemia (CLL) [33–35] and in MM [36, 37].
Multiple myeloma
MM was the first hematological malignancy, in which increased angiogenesis rate was detected [21, 38]. MM is characterized by proliferation of malignant plasma cells that accumulate in the bone marrow and often produce a monoclonal immunoglobulin. New vessel formation in the bone marrow seems to play an important role in the pathogenesis of MM [39, 40]. Increased bone marrow microvessel density (MVD) in patients with MM appears to be also an important prognostic factor [41]. Malignant plasma cells can secrete various cytokines, including VEGF, basic fibroblast growth factor (bFGF), and hepatocyte growth factor (HGF), all known for their pro-angiogenic activity [42]. It has been shown that MM cells are capable of secreting VEGF in response to Interleukin-6 (IL-6) stimulation; in response to that VEGF stimulation microvascular endothelial cells and bone marrow stromal cells secrete in turn IL-6, a potent growth factor for malignant plasma cells, thus closing a paracrine loop [43]. Specifically, increased microvessel density (MVD) in the BM of MM patients has been correlated with disease progression and poor prognosis [21, 23]. Moreover, VEGF also exerts direct effects on MM cell migration, proliferation, survival, and drug resistance. VEGF triggered effects in MM cells are predominantly mediated via VEGFR 1 and in endothelial cells, predominantly via VEGF R2 [44]. Rajkumar et al. showed a gradual increase of bone marrow angiogenesis along the disease spectrum from monoclonal gammopathy of undetermined significance (MGUS) to smoldering MM, newly diagnosed MM and relapsed MM [45], though the expression levels of VEGF, bFGF, and their receptors were similar among MGUS, smoldering MM, and newly diagnosed MM [46], rising the hypothesis that MVD increase in plasma cell neoplasias could be rather a function of chronology.
Acute leukemias
The first demonstration that leukemia progression might be accompanied by an increase of bone marrow vascularization was provided by Judah Folkman's group [47]. In their studies, it was demonstrated that the bone marrow of acute lymphoblastic leukemia (ALL) patients had increased blood vessel content, compared to normal counterparts. Moreover, it was also shown that urine and peripheral blood samples from ALL patients contained elevated levels of pro-angiogenic growth factors, namely bFGF and VEGF, which correlated with the increase of bone marrow angiogenesis [48]. The existence of an "angiogenesis switch", first proposed for solid tumors [49], was therefore suggested to apply to hematological malignancies as well. "Angiogenesis switch" in leukemia is documented by increased bone marrow MVD, increased expression of HIF-1, multiple pro-angiogenic factors (VEGF, bFGF, angiopoietin-2), soluble VEGFR, and decreased expression of endogenous angiogenesis inhibitors, such as thrombospondin-1 [50, 51].
In a recent study by Norén-Nyström et al. [52] MVD, analyzed on 185 bone marrow biopsies, was higher in T-ALL compared to B-ALL. In the B-ALL group, cases with t(12;21) were characterized by a low MVD, while patients with hyperdiploid leukemia showed a high MVD. Similarly, in previously untreated acute myeloid leukemia (AML), increased levels of plasma VEGF correlate with reduced survival and lower remission rates [53]. In addition to that, isolated blast cells from leukemia patients also produce VEGF and the cellular level of VEGF in AML patients has been identified as independent prognostic risk factor [28]. In a reccent study [54] dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) was used as a non-invasive technique to measure bone marrow angiogenesis in AML. DCE-MRI was performed beforte treatment and on day 7 after induction chemotherapy. Thereby, bone marrow angiogenesis with remission, rate overall and disease-free survival.
Myeloproliferative neoplasms
The available data on angiogenesis and expression of VEGF and its receptors in the bone marrow of patients with
Anti-angiogenic therapies in hematological malignancies
Anti-angiogenic therapies are mostly based on inhibiting the binding of VEGF to VEGFR by neutralizing antibodies to the ligand or to the receptor, soluble receptors, small molecule inhibitors or are directed against the tyrosine kinase activity of the VEGF receptors (Figure 1). The first anti-angiogenic agent to be approved in solid tumors was bevacizumab (Avastin™, Genentech), a humanized anti-VEGF monoclonal antibody. Administration of bevacizumab, in combination with cytotoxic chemotherapy, conferred benefits to patients with metastatic colorectal cancer, non-squamous, non-small cell lung cancer and metastatic breast cancer [62–64]. Additionally, two small-molecule inhibitors targeting VEGFRs and other kinases, sorafenib (Nexavar™, Bayer and Onyx pharmaceuticals) and sunitinib (Sutent™, Pfizer), have been approved based on their efficacy in treating renal cell- and hepatocellular carcinoma [65, 66]. A growing list of anti-angiogenics is now available, either in various stages of clinical development or as components of standard clinical regimens. The major classes of anti-angiogenic therapy include: (1) direct anti-VEGF acting molecules (anti-VEGF antibodies,
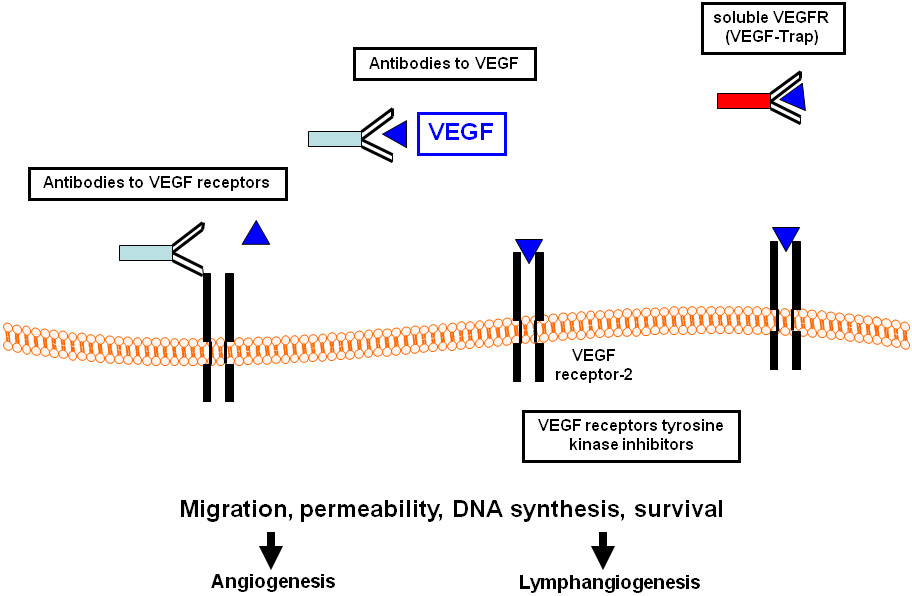
Figure 1
Figure 1 caption
Therapeutic strategies to target the VEGF/VEGF receptor system. VEGF, vascular endothelial growth factor.
In our review, we will focus on several molecules interfering with the VEGF/VEGFR system, which already have been approved or are currently evaluated in clinical trials for treatment of hematological malignancies (Table 1).
Table 1
Selection of clinical trials and approved anti-angiogenic therapies in hematological malignancies
| Drug | Target | Study entities | Approved for |
|---|---|---|---|
| Receptor tyrosine kinase inhibitors | |||
| PTK787/ZK 222584 (Vatalanib®) | VEGFR1-3, PDGFRβ, c-Kit | AML, PMF, MDS, CML, DLBCL, MM | |
| SU5416 | VEGFR1-2, c-kit, Flt3 | AML, MDS, MM, MPN | |
| Sorafenib (Nexavar®) | VEGFR2-3, B-Raf, Faf-1, PDGFRβ | AML, ALL, MDS, CML, CLL, NHL, MM | Advanced renal cell carcinoma, HCC |
| Sunitinib (Sutent®) | VEGFR1-3, PDGFRα+β, c-kit, Flt3 | AML, MDS, CLL, Myeloma, NHL | Advanced renal cell carcinoma, GIST |
| PKC-412 (Midostaurin) | VEGFR2, PKC, PDGFR, Flt3, c-Kit | AML | |
| Cediranib (Recentin®) | VEGFR1-3, PDGFRβ, c-Kit | AML, MDS, CLL | |
| Proteasome inhibitors | |||
| Bortezomib (Velcade®) | 26S proteasome, NF-κB | AML, ALL, MDS, CML, NHL, MCL | MM, MCL |
| Anti-VEGF strategies | |||
| Bevacizumab (Avastin®) | VEGF-A | AML, MDS, CLL, CML, NHL, MM | Metastatic colorectal cancer, NSCLC, breast cancer |
| Immunomodulatory drugs | |||
| Thalidomide | bFGF, VEGF, IL-6 | AML, MDS, MPN, CLL, NHL, MM | MM |
| Lenalidomide (Revlimid®) | bFGF, VEGF, IL-6 | AML, MDS, CLL, NHL | MM, 5q- MDS |
Anti-VEGF monoclonal antibodies
Conclusions and future directions
Angiogenic and especially VEGF/VEGFR pathways are involved in the pathophysiology of hematological malignancies including multiple myeloma, acute and chronic leukemias, MPN and lymphomas. Although VEGF/VEGFR-related pathways seems to be the most relevant regulators of neoangiogenesis, vasculogenesis and recruitment of endothelial progenitor cells in such instances, but other pathways are important too. Further, VEGF/VEGFR interactions can stimulate proliferation, migration and survival of leukemia/lymphoma cells by autocrinous and paracrinous loops. Novel agents, targeting VEGF, its receptors, and other angiogenic pathways, are in various stages of clinical development and investigation in hematological malignancies. As we know from the the treatment of solid tumors, combination therapies of different anti-angiogenic molecules with chemotherapy or irradiation increases treatment efficacy. Especially, as blocking VEGF activity has been shown to sensitize the vasculature and improve the delivery of cytotoxic drugs to tumor and endothelial cells. However, not all patients treated with anti-angiogenic therapies benefit from this kind of therapy and in most cases, the effect is transient. Therefore, there is an urgent need for biomarkers to identify patients likely to benefit from anti-angiogenic treatments, to select the optimal dose to minimize side effects, and to understand the mechanisms of resistance. Preclinical models suggest multiple mechanisms involved in acquired or primary resistance against anti-angiogenic therapies. Finally, also these "targeted therapies" has side effects profiles which must be considered carefully.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2
References
- Angiogenesis in cancer, vascular, rheumatoid, and other disease. Nat Med. 1995;1:27-31.
- Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-1186.
- Impaired recruitment of bone-marrow derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med. 2001;7:1194-1201.
- The biology of VEGF and its receptors. Nat Med. 2003;9:669-676.
- Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276:3222-3230.
- Vascular endothelial growth factor and angiopoietin-1 stimulate postnatal hematopoiesis by recruitment of vasculogenic and hematopoietic stem cells. J Exp Med. 2001;193:1005-1014.
- Expression of vascular endothelial growth factor and its receptors in hematopoietic malignancies. Cancer Res. 1999;59:728-733.
- Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167-174.
- Autocrine-paracrine VEGF loops potentiate the maturation of megakaryocytic precursors through Flt1 receptor. Blood. 2003;101:1316-1323.
- The delta paradox: DLL4 blockade leads to more tumour vessels but less tumour growth. Nat Rev Cancer. 2007;7:327-331.
- Delta-like 4/Notch signaling and its therapeutic implications. Clin Cancer Res. 2007;13:7243-7246.
- Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp Cell Res. 2007;313:1-9.
- Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032-1037.
- Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083-1087.
- Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res. 2007;67:11244-11253.
- The role of angiogenesis in the biology and therapy of myelodysplastic syndromes. Curr Hematol Rep. 2004;3:184-191.
- Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2-V617F mutational status. Br J Haematol. 2009;146:150-157.
- Increased angiogenesis in the bone marrow of patients with acute myeloid leukemia. Blood. 2000;95:2637-2644.
- Angiogenesis in myelodysplastic syndromes. Br J Cancer. 1999;81:1398-1401.
- Angiogenesis in nodal B cell lymphomas: a high throughput study. J Clin Pathol. 2007;60:476-482.
- Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87:503-508.
- Analysis of concerted expression of angiogenic growth factors in acute myeloid leukemia: expression of angiopoietin-2 represents an independent prognostic factor for overall survival. J Clin Oncol. 2005;23:1109-1117.
- Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase2 secretion parallel progression of human multiple myeloma. Blood. 1999;93:30643073-.
- Numerous growth factors, cytokines, and chemokines are secreted by human CD34(+) cells, myeloblasts, erythroblasts, and megakaryoblasts and regulate normal hematopoiesis in an autocrine/paracrine manner. Blood. 2001;97:3075-3085.
- Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306-1309.
- Vascular endothelial growth factor, a possible paracrine growth factor in human acute myeloid leukemia. Blood. 1997;89:1870-1875.
- Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511-521.
- Cellular vascular endothelial growth factor is a predictor of outcome in patients with acute myeloid leukemia. Blood. 1999;94:3717-3721.
- The Notch signaling pathway: transcriptional regulation at Notch target genes. Cell Mol Life Sci. 2009;66:1631-1646.
- Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269-271.
- Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood. 2002;99:3398-3403.
- Aberrant expression of Notch1 interferes with the B-lymphoid phenotype of neoplastic B cells in classical Hodgkin lymphoma. Leukemia. 2008;22:1587-1594.
- Notch2 is involved in the overexpression of CD23 in B-cell chronic lymphocytic leukemia. Blood. 2002;99:3742-3747.
- Induction of apoptosis by proteasome inhibitors in B-CLL cells is associated with downregulation of CD23 and inactivation of Notch2. Leukemia. 2005;19:260-267.
- Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113:856-865.
- Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004;103:3503-3510.
- Jagged1-induced Notch signaling drives proliferation of multiple myeloma cells. Blood. 2004;103:3511-3515.
- Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006;66:184-191.
- Bone marrow microvessel density is a prognostic factor for survival in patients with multiple myeloma. Ann Hematol. 2000;79:574-577.
- Prognostic value of bone marrow angiogenesis in multiple myeloma. Clin Cancer Res. 2000;6:3111-3116.
- Increased bone marrow microvessel density in newly diagnosed multiple myeloma carries a poor prognosis. Semin Oncol. 2001;28:565-569.
- Expression of VEGF and its receptors by myeloma cells. Leukemia. 2003;17:2025-2031.
- Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood. 2001;98:428-435.
- The pathophysiological role of VEGF in hematological malignancies: Therapeutic implications. Blood. 2005;105:138395-.
- A review of angiogenesis and anti-angiogenic therapy in hematologic malignancies. J Hematother Stem Cell Res. 2002;11:33-47.
- Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: evidence favoring loss of marrow angiogenesisinhibitory activity with disease progression. Blood. 2004;104:1159-1165.
- Spectrum of tumor angiogenesis in the bone marrow of children with acute lymphoblastic leukemia. Am J Pathol. 1997;150:815-821.
- Clinical importance of serum vascular endothelial and basic fibroblast growth factors in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2001;42:83-88.
- Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353-364.
- Angiogenesis and antiangiogenic therapy in hematologic malignancies. Crit Rev Oncol/Hematol. 2007;62:105-118.
- Dysregulated angiogenesis in B-chronic lymphocytic leukemia: morphologic, immunohistochemical, and flow cytometric evidence. Diagn Pathol. 2008;3:16-.
- Vascular density in childhood acute lymphoblastic leukaemia correlates to biological factors and outcome. Br J Haematol. 2009;146:521-350.
- Plasma vascular endothelial growth factor levels have prognostic significance in patients with acute myeloid leukemia but not in patients with myelodysplastic syndromes. Cancer. 2002;95:1923-1930.
- Changes in MR bone marrow angiogenesis on day 7 after induction chemotherapy can predict outcome of acute myeloid leukemia. Haematologica. 2010.
- Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. Am J Pathol. 2000;157:15-19.
- Increased expression of vascular endothelial growth factor (VEGF) in bone marrow of patients with myeloproliferative disorders (MPD). Pathol Oncol Res. 2003;9:170-173.
- VEGF expression correlates with microvessel density in Philadelphia chromosome-negative chronic myeloproliferative disorders. Am J Clin Pathol. 2007;128:966-973.
- Chronic idiopathic myelofibrosis: independent prognostic importance of bone marrow microvascular density evaluated by CD105 (endoglin) immunostaining. Mod Pathol. 2004;17:1513-1520.
- A gain-of-function mutation of JAK2 in myeloproliferative isorders. N Engl J Med. 2005;352:1779-1790.
- Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377-1380.
- Evaluation of circulating endothelial cells, VEGF and VEGFR2 serum levels in patients with chronic myeloproliferative diseases. Hematol Oncol. 2008;26:235-239.
- Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-2342.
- Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542-2550.
- Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666-2676.
- Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125-134.
- Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115-124.
- Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593-4599.
- Bevacizumab reduces VEGF expression in patients with relapsed and refractory acute myeloid leukemia without clinical antileukemic activity. Leukemia. 2007;21:1310-1312.
- Targeting vascular endothelial growth factor for relapsed and refractory adult acute myelogenous leukemias: therapy with sequential 1-beta-d-arabinofuranosylcytosine, mitoxantrone, and bevacizumab. Clin Cancer Res. 2004;10:3577-3585.
- A phase 2 clinical study of SU5416 in patients with refractory acute myeloid leukemia. Blood. 2003;102:2763-2767.
- Stable remission after administration of the receptor tyrosine kinase inhibitor SU5416 in a patient with refractory acute myeloid leukemia. Blood. 2001;98:241-243.
- SU5416, a small molecule tyrosine kinase receptor inhibitor, has biologic activity in patients with refractory acute myeloid leukemia or myelodysplastic syndromes. Blood. 2003;102:795-801.
- PTK787/ZK 22 a specific vascular endothelial growth factor-receptor tyrosine kinase inhibitor, affects the anatomy of the tumor vascular bed and the functional vascular properties as detected by dynamic enhanced magnetic resonance imaging. Cancer Res. 2584;62:4015-4022.
- Phase I clinical and pharmacokinetic study of PTK/ZK, a multiple VEGF receptor inhibitor, in patients with liver metastases from solid tumors. Eur J Cancer. 2005;41:1291-1299.
- PTK787/ZK 22 a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2584;60:2178-2189.
- The vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584 inhibits growth and migration of multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2002;62:5019-5026.
- Phase 1 study of PTK787/ZK 22 a small molecule tyrosine kinase receptor inhibitor, for the treatment of acute myeloid leukemia and myelodysplastic syndrome. Leukemia. 2584;20:952-957.
- Additive effect of PTK787/ZK 22 a potent inhibitor of VEGFR phosphorylation, with Idarubicin in the treatment of acute myeloid leukemia. Exp Hematol. 2584;37:679-691.
- PTK787/ZK 22 a small molecule tyrosine kinase receptor inhibitor of vascular endothelial growth factor (VEGF), has modest activity in myelofibrosis with myeloid metaplasia. Leuk Res. 2584;31:891-897.
- AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389-4400.
- Phase I clinical study of AZD an oral vascular endothelial growth factor signaling inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2171;20:3045-3054.
- An open-label, phase I study of cediranib (RECENTIN) in patients with acute myeloid leukemia. Leuk Res. 2010;34:196-202.
- Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565-1571.
- Extended survival in advanced and refractory multiple myeloma after single-agent thalidomide: identification of prognostic factors in a phase 2 study of 169 patients. Blood. 2001;98:492-494.
- Thalidomide-effect on T cell subsets as a possible mechanism of action. Int J Lepr Other Mycobact Dis. 1985;53:201-205.
- Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91:4082-4085.
- Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15:1950-1961.
- Properties of thalidomide and its analogues: implications for anticancer therapy. AAPS J. 2005;7:E14-19.
- Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br J Cancer. 2002;87:1166-1172.
- Phase III clinical trial of thalidomide plus dexamethasone compared with dexamethasone alone in newly diagnosed multiple myeloma: a clinical trial coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol. 2006;24:431-436.
- Single agent thalidomide in patients with relapsed or refractory acute myeloid leukaemia. Br J Haematol. 2003;123:436-441.
- Efficacy and safety of thalidomide in patients with acute myeloid leukemia. Blood. 2002;99:834-839.
- Combination of 5-azacytidine and thalidomide for the treatment of myelodysplastic syndromes and acute myeloid leukemia. Cancer. 2008;113:1596-1604.
- Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006;106:1974-1984.
- Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol. 2009;2:36-.
- Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123-2132.
- Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133-2142.
- Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108:1158-1164.
- Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27:4760-4766.
- Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res. 2001;7:1419-1428.
- Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Research. 2006;66:184-191.
- United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clinical Cancer Research. 2006;12:2955-2960.
- Bortezomib Plus Melphalan and Prednisone Compared With Melphalan and Prednisone in Previously Untreated Multiple Myeloma: Updated Follow-Up and Impact of Subsequent Therapy in the Phase III VISTA Trial. J Clin Oncol. 2010;28:2259-2266.
- Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010.
- Phase I and pharmacokinetic study of bortezomib in combination with idarubicin and cytarabine in patients with acute myelogenous leukemia. Clin Cancer Res. 2008;14:1446-1454.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.