Eicosapentaenoic acid inhibits endothelial cell migration in vitro
Journal of Angiogenesis Research. 2010;
Received: 1 April 2010 | Accepted: 9 July 2010 | Published: 9 July 2010
Vascular Cell ISSN: 2045-824X
Abstract
Background
As
Methods
To this purpose, using functional and morphological
Results
We report here that incubation of endothelial cells with
Conclusions
Given the importance of endothelial cell migration in the repair of vascular injuries, these
Background
Dietary intake of
In this study, we focused on the effect of
Furthermore,
Here, we surmised that, in addition to these responses, PUFA might interfere with other biological processes that involve membrane proteins, such as cell migration.
Results
Addition of n-3 PUFA increases the membrane content of EPA and DHA
Incubation of confluent EC (for 24 hours at 37°C) with a mixture of
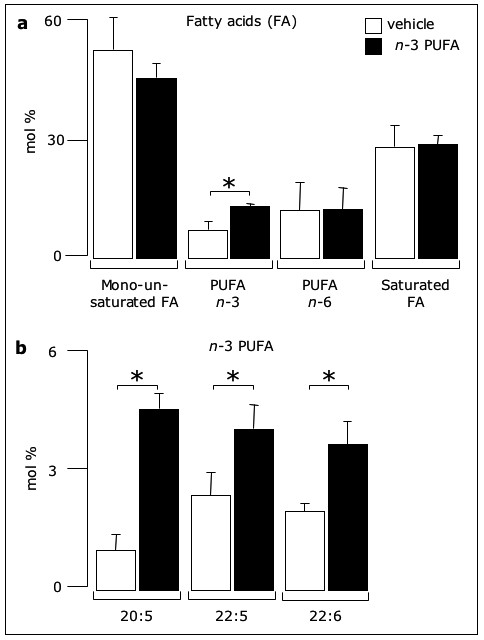
Figure 1
Figure 1 caption
-3 PUFA change the relative fatty acid composition of endothelial membranes. Confluent EC were incubated for 24 hours with either ethanol () or 50 μM -3 PUFA EE. Then, membranes were analyzed by chromatography for the composition of (a) total -3 PUFA, -6 PUFA and saturated fatty acids, as well as (b) the -3 PUFA 20:5, 22:5 and 22:6. Results are mean ± SD from three experiments (* p < 0.05).
n-3 PUFA inhibit endothelial cell migration
We next examined the effect of
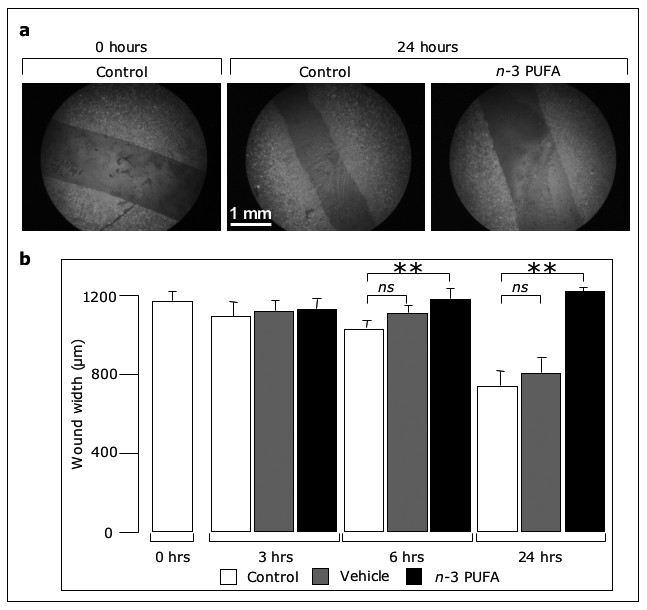
Figure 2
Figure 2 caption
-3 PUFA inhibit migration into a wound. Confluent EC were wounded and incubated with medium, vehicle or -3 PUFA EE. Then, cells were fixed, stained with FITC-phalloidin and examined by fluorescence microscopy. Representative samples are shown in (a), while the wound widths are reported in (b) as mean ± SD from three experiments performed in duplicate (** p < 0.01).
EPA is responsible for the n-3 PUFA effect on cell migration
The mixture of
Table 1
Specificity of the
| Medium | 852 ± 15 |
| Vehicle (ethanol) | 905 ± 118 |
| PUFA EE | 1166 ± 94 ** |
| DHA EE | 896 ± 51 |
| EPA EE | 1189 ± 60 ** |
| DHA EE + EPA EE | 1156 ± 9 ** |
As above, at 24 hours, the wound width was reduced in control- and vehicle (ethanol)-treated cells, but not in cells treated with the mixture of
n-3 PUFA-dependent inhibition of migration is MAPK-independent
Mitogen-Activated Protein Kinases (MAPK) are key regulators of cell migration [12]. In particular, EC migration into a wound requires ERK-1/2 activation [13]. In agreement with these findings, we observed that migration was reduced in cells treated with the ERK-1/2 inhibitor PD098059, compared with control-treated cells (Fig. 3a). However, when analyzing cells that had been treated in either the presence or absence of the
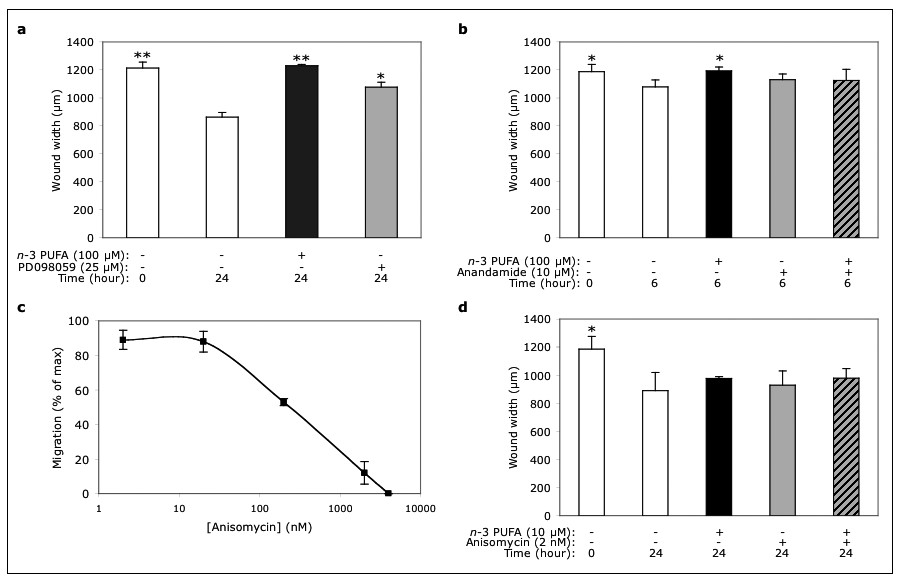
Figure 3
Figure 3 caption
Role of MAPK in the-3 PUFA-dependent inhibition of migration. At 24 hours after plating, EC monolayers were switched to low-serum (1%) medium and pre-treated with the indicated MAPK regulators for additional 24 hours. Finally, monolayers were wounded and incubated (in low-serum medium) in either the absence or presence of the indicated compounds for either 6 (b) or 24 (a, c and d) hours. Data are mean ± SD from three experiments, each performed in duplicate (* p < 0.05; ** p < 0.01, compared with the control treatment at the same time point, ).
In addition, we found that the p38/SAPK2 activator anisomycin induced dose-dependent (2-2,000 nM) inhibition of migration, with half-maximal inhibition at about 250 nM (Fig. 3c). However, non-inhibitory concentrations of
n-3 PUFA-dependent inhibition of migration is associated with cytoskeletal changes
Fluorescence microscopy revealed morphological differences in the migrating cells that were facing the wound (Fig. 4a). Specifically, in control-and vehicle-treated cells, vinculin-containing focal adhesions (FA) distributed evenly at the base of the leading lamella, while actin filaments aligned in parallel to the wound. In contrast, in
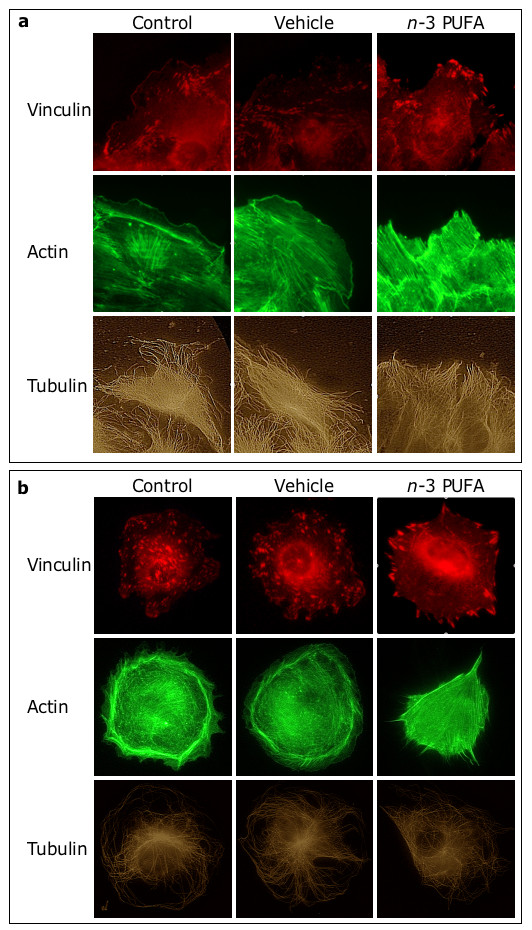
Figure 4
Figure 4 caption
-3 PUFA affect FA and actin filaments. In (a), EC were grown to confluence and wounded. In (b), EC were allowed to adhere (as non-contacting cells) for 2 hours. Then, cells were incubated for 24 hours (a) or 1 hour (b) with medium (), ethanol () and 100 μM -3 PUFA EE.
Similar cytoskeletal changes were detectable in the confluent cells that were not facing the wound (not shown) and in non-contacting cells (Fig. 4b). Specifically, in non-contacting cells incubated with medium or vehicle, FA evenly distributed throughout the cell, while actin filaments organized in cortical fibers. In contrast, upon treatment with
We also determined that the fraction of non-contacting cells with peripheral FA was negligible in control- and vehicle-treated cells (0.0 ± 0.0 and 0.7 ± 1.2%, respectively), but high (70.0 ± 10.6%) in cells treated with the mixture of
Discussion
The major findings of this study are that
The anti-migratory effect of EPA was not expression of a nonspecific anti-motility action. Actually, using the modified Boyden chamber assay, previous studies reported that EPA (but not DHA) increased EC migration [14] and rescued the anti-migratory action of cholesterol [15]. These studies are not at odds with the herein reported inhibitory action of EPA, as the Boyden assay analyzes the individual migration of single cells, while the wound assay evaluates the collective migration of cohesive cell sheets. In addition, our findings mirror the observation that EPA (but not DHA) inhibited the ability of EC to form tube-like structures in collagen gels, which requires coordinated motility and cytoskeletal rearrangements of cohesive EC [16].
Published evidence prompted us to evaluate the possible involvement of the MAPK. In particular, hydrogen peroxide-dependent distribution of the FA to the cell periphery [17] (similar to the one induced by
Most of our experiments relied on the use of a mixture of
However, we also used purified
Other studies have shown differences in the biological effects of EPA and DHA [25]. In addition to differences in metabolic and hemodynamic effects (
Clearly,
Finally, besides migration, one should take into account other effects of the
Conclusions
In conclusion, we have reported here that
Methods
Cell lines, antibodies and reagents
The endothelial cell line H5V, which was derived from the murine heart [37], was cultured in D-MEM (Gibco-BRL) supplemented with 10% fetal bovine serum (Sigma). Mouse anti-vinculin mAb hVIN-1, mouse anti-alpha-tubulin mAb B-5-1-2 and FITC-phalloidin were from Sigma. TRITC-labeled anti-IgG (H+L) anti-mouse antibody was from Jackson Immuno Research Laboratories Inc. The mixture of
Fatty acid analysis
Membrane isolation and analysis of their relative fatty acid composition were performed as described [38]. Briefly, lipids from approximately 2.5 × 105 EC were extracted according to the method of Folch [39]. Then, aliquots of the chloroform phases containing lipids were evaporated to dryness. Fatty acid methyl esters in total lipids were prepared and injected into an Autosystem XL gas chromatograph (Perkin Elmer) connected with a flame ionisation detector. Individual fatty acid methyl esters were identified by comparing their retention times with corresponding standards (run in parallel), and their composition was calculated using a Turbochrom system, version 6.1 (Perkin Elmer).
Wound assay
Cells were seeded (at a density of 1.5 × 105/cm2) onto glass cover slips (that had been coated with fibronectin; 7 μg/mL) and grown to confluence. Then, the culture medium was removed, and scratch wounds were produced using a plastic tip for 1,000 μL pipettes. After two washes with D-PBS, cells were incubated with D-MEM medium containing 10% serum, in the presence of either medium alone, or the vehicle (ethanol) or
Immunofluorescence microscopy
For the analysis of non-contacting cells, cells were seeded onto fibronectin-coated glass cover slips, allowed to adhere for 120 minutes and then incubated with the indicated compound for additional 60 minutes. Cells were then treated as above and stained with primary and TRITC-labeled secondary antibodies, as described [41]. Cover slips were mounted in 488-Mowiol and analyzed with a Zeiss Axiophot microscope (equipped with 100X Plan-Neofluar Ph3 objective lens). A similar protocol was used for analyzing migrating cells at the wound edge.
Acknowledgements
This work was partially supported by Società Prodotti Antibiotici, Milano.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2
Authors’ original file for figure 3
Authors’ original file for figure 4
Authors’ original file for figure 5
References
- Effect of different antilipidemic agents and diets on mortality: a systematic review. Arch Intern Med. 2005;165(7):725-730.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Lancet. 1999, 354 (9177): 447-455. 10.1016/S0140-6736(99)07072-5.
- The biochemistry of n-3 polyunsaturated fatty acids. J Biol Chem. 2002;277(11):8755-8758.
- Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J Biol Chem. 2000;275(1):261-270.
- Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J Biol Chem. 2001;276(40):37335-37340.
- Long-chain n-3 fatty acids in lipid rafts: implications for anti-inflammatory effects. J Cardiovasc Med (Hagerstown). 2007;8(Suppl 1):S30-33.
- Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1(1):31-39.
- N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr Opin Lipidol. 2008;19(3):242-247.
- Docosahexaneoic acid (22:6,n-3) regulates rat hepatocyte SREBP-1 nuclear abundance by Erk-and 26 S proteasome-dependent pathways. J Lipid Res. 2006;47(1):181-192.
- Omega-3 fatty acids and endothelial leukocyte adhesion molecules. Prostaglandins Leukot Essent Fatty Acids. 1995;52(2-3):191-195.
- The omega-3 fatty acid docosahexaenoate attenuates endothelial cyclooxygenase-2 induction through both NADP(H) oxidase and PKC epsilon inhibition. Proc Natl Acad Sci USA. 2006;103(41):15184-15189.
- MAP kinases and cell migration. J Cell Sci. 2004;117(Pt 20):4619-4628.
- Lack of ERK activation and cell migration in FGF-2-deficient endothelial cells. Faseb J. 2002;16(6):598-600.
- Enhancement of migration in bovine endothelial cells by eicosapentaenoic acid pretreatment. Atherosclerosis. 1991;87(1):57-64.
- Docosapentaenoic acid (22:5, n-3), an elongation metabolite of eicosapentaenoic acid (20:5, n-3), is a potent stimulator of endothelial cell migration on pretreatment in vitro. Prostaglandins Leukot Essent Fatty Acids. 1996;54(5):319-325.
- Eicosapentaenoic acid inhibits tube formation of vascular endothelial cells in vitro. Lipids. 1991;26(4):271-276.
- Hydrogen peroxide-induced endothelial retraction is accompanied by a loss of the normal spatial organization of endothelial cell adhesion molecules. Am J Pathol. 1995;147(3):627-641.
- Extracellular signal-regulated kinase mediates phosphorylation of tropomyosin-1 to promote cytoskeleton remodeling in response to oxidative stress: impact on membrane blebbing. Mol Biol Cell. 2003;14(4):1418-1432.
- SAPK2/p38-dependent F-actin reorganization regulates early membrane blebbing during stress-induced apoptosis. J Cell Biol. 1998;143(5):1361-1373.
- Docosahexaenoic acid induces apoptosis in proliferating human endothelial cells. J Cell Physiol. 2005;204(3):881-888.
- Eicosapentaenoic acid and docosahexaenoic acid modulate MAP kinase (ERK1/ERK2) signaling in human T cells. J Lipid Res. 2001;42(12):2015-2020.
- Differential effects of low-dose docosahexaenoic acid and eicosapentaenoic acid on the regulation of mitogenic signaling pathways in mesangial cells. J Lab Clin Med. 2003;141(5):318-329.
- Effect of dietary supplementation with omega-3 fatty acids on progression of atherosclerosis in carotid arteries. Cardiovasc Res. 2002;54(1):183-190.
- Polyunsaturated fatty acids inhibit T cell signal transduction by modification of detergent-insoluble membrane domains. J Cell Biol. 1998;143(3):637-644.
- The independent effects of eicosapentaenoic acid and docosahexaenoic acid on cardiovascular risk factors in humans. Curr Opin Clin Nutr Metab Care. 2006;9(2):95-104.
- Fatty acid modulation of endothelial activation. Am J Clin Nutr. 2000;71(1 Suppl):213S-223S.
- Effects of purified eicosapentaenoic acid and docosahexaenoic acid on platelet, fibrinolytic and vascular function in hypertensive type 2 diabetic patients. Atherosclerosis. 2003;166(1):85-93.
- Inhibition of endothelial cell regrowth. Cessation of aortic endothelial cell replication after balloon catheter denudation. Arteriosclerosis. 1982;2(3):216-220.
- Mechanisms of neointima formation--lessons from experimental models. Vasc Med. 1997;2(3):179-189.
- Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105(25):3017-3024.
- Mobilized endothelial progenitor cells by granulocyte-macrophage colony-stimulating factor accelerate reendothelialization and reduce vascular inflammation after intravascular radiation. Circulation. 2003;108(23):2918-2925.
- Wound healing--aiming for perfect skin regeneration. Science. 1997;276(5309):75-81.
- The effects of omega-3 fatty acid diet enrichment on wound healing. Vet Dermatol. 1999;10:283-290.
- Omega-3 fatty acids effect on wound healing. Wound Repair Regen. 2008;16(3):337-345.
- Influence of topical administration of n-3 and n-6 essential and n-9 nonessential fatty acids on the healing of cutaneous wounds. Wound Repair Regen. 2004;12(2):235-243.
- Basic mechanisms behind the effects of n-3 fatty acids on cardiovascular disease. Prostaglandins Leukot Essent Fatty Acids. 2008;79(3-5):109-115.
- Progressive growth in immunodeficient mice and host cell recruitment by mouse endothelial cells transformed by polyoma middle-sized T antigen: implications for the pathogenesis of opportunistic vascular tumors. Proc Natl Acad Sci USA. 1994;91(15):7291-7295.
- Early modifications of fatty acid composition in plasma phospholipids, platelets and mononucleates of healthy volunteers after low doses of n-3 polyunsaturated fatty acids. Eur J Clin Pharmacol. 2004;60(3):183-190.
- A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497-509.
- Expression of junctional adhesion molecule-A prevents spontaneous and random motility. J Cell Sci. 2005;118(Pt 3):623-632.
- Opposite effects of tumor necrosis factor and soluble fibronectin on junctional adhesion molecule-A in endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2005;288(6):L1081-1088.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.