Fine mapping of the hereditary haemorrhagic telangiectasia (HHT)3 locus on chromosome 5 excludes VE-Cadherin-2, Sprouty4 and other interval genes
Journal of Angiogenesis Research. 2010;
Received: 8 June 2010 | Accepted: 11 August 2010 | Published: 11 August 2010
Vascular Cell ISSN: 2045-824X
Abstract
Background
There is significant interest in new loci for the inherited condition hereditary haemorrhagic telangiectasia (HHT) because the known disease genes encode proteins involved in vascular transforming growth factor (TGF)-β signalling pathways, and the disease phenotype appears to be unmasked or provoked by angiogenesis in man and animal models. In a previous study, we mapped a new locus for HHT (
Methods
Extended analyses in the interval-defining pedigree were performed using informative genomic sequence variants identified during candidate gene sequencing. These variants were amplified by polymerase chain reaction; sequenced on an ABI 3730xl, and analysed using FinchTV V1.4.0 software.
Results
Informative genomic sequence variants were used to construct haplotypes permitting more precise citing of recombination breakpoints. These reduced the uninformative centromeric region from 141.2-144 Mb to between 141.9-142.6 Mb, and the uninformative telomeric region from 145.2-146.9 Mb to between 146.1-146.4 Mb.
Conclusions
The
Background
Transforming growth factor (TGF)-β superfamily signalling is of fundamental importance to developmental and physiological regulation. In these pathways (reviewed in [1, 2]), ligands such as TGF-βs, bone morphogenetic proteins (BMP)s, activins, nodals, growth/differentiation factors (GDF)s and inhibins bind to receptor complexes of paired type I and type II transmembrane receptor serine/threonine kinases. Activated type I receptors (ALKs 1-7) phosphorylate receptor-associated Smad proteins in complex-specific patterns [3–5]. There is increasing recognition of the role of alternative signalling pathways for particular ligands within designated cell types. In endothelial cells (ECs), signalling through the TGF-β type II receptor, TβRII, can be propagated not only through ALK-5 via SMAD2/3 as in other cell types, but also through ALK-1 via SMAD1/5/8, providing two mutually antagonistic pathways [6, 7]. The transmembrane glycoprotein endoglin is an accessory TGF-β receptor, highly expressed on ECs, and is one factor modulating the balance between ALK-1 and ALK-5 pathways [8].
The inherited vascular condition hereditary haemorrhagic telangiectasia (HHT) [9] is of significant relevance to TGF-β signalling because the genes for endoglin, ALK-1 and SMAD4 (a co-Smad and downstream effector of the TGF-β signalling pathway), are mutated in different HHT families [10–12]. HHT is transmitted as an autosomal dominant trait due to a single mutation in either
HHT serves not only as a vascular model of aberrant TGF-β superfamily signalling, but also as a model of aberrant angiogenesis [13, 14]. The abnormal blood vessels develop only in selected vascular beds (telangiectasia particularly in mucocutaneous and gastrointestinal sites; arteriovenous malformations (AVMs) most commonly in pulmonary, hepatic and cerebral circulations) [9, 15]. At each site, only a small proportion of vessels are abnormal. The context in which HHT mutations are deleterious, when allowing apparently normal endothelial function for most vessels, now appear to be angiogenic in origin. Early studies modelling HHT in transgenic animals provided evidence of aberrant angiogenesis. Heterozygous mice developed HHT-like features;
A current model to explain these observations, discussed in more detail in [26], is based on the EC-mural cell axis defined by Sato and Rifkin [27]. In angiogenesis, HHT mutations (endoglin and ALK-1) appear to impair recruitment of mural cells to the angiogenic sprout [7, 28] at least in part via reduced EC secretion of TGF-β1 [29, 30] and/or reduced TGF-β1 induced responses [7, 29] resulting in defective mural cell stabilisation of the nascent vessel and persistent, excessive, EC proliferation. Thalidomide, which induced vessel maturation in Eng+/- mice which normally suffer from excessive angiogenesis, appears to target mural cell recruitment, by increasing endothelial expression of PDGF-B at the endothelial tip cell, thus facilitating recruitment of pericytes that express PDGFR-b, associated with increasing pericyte proliferation [25].
Further HHT genes were therefore predicted to identify new components or regulators of TGF-β/BMP signalling pathways of particular relevance to angiogenesis. More than 80% of HHT patients carry a sequence variation in
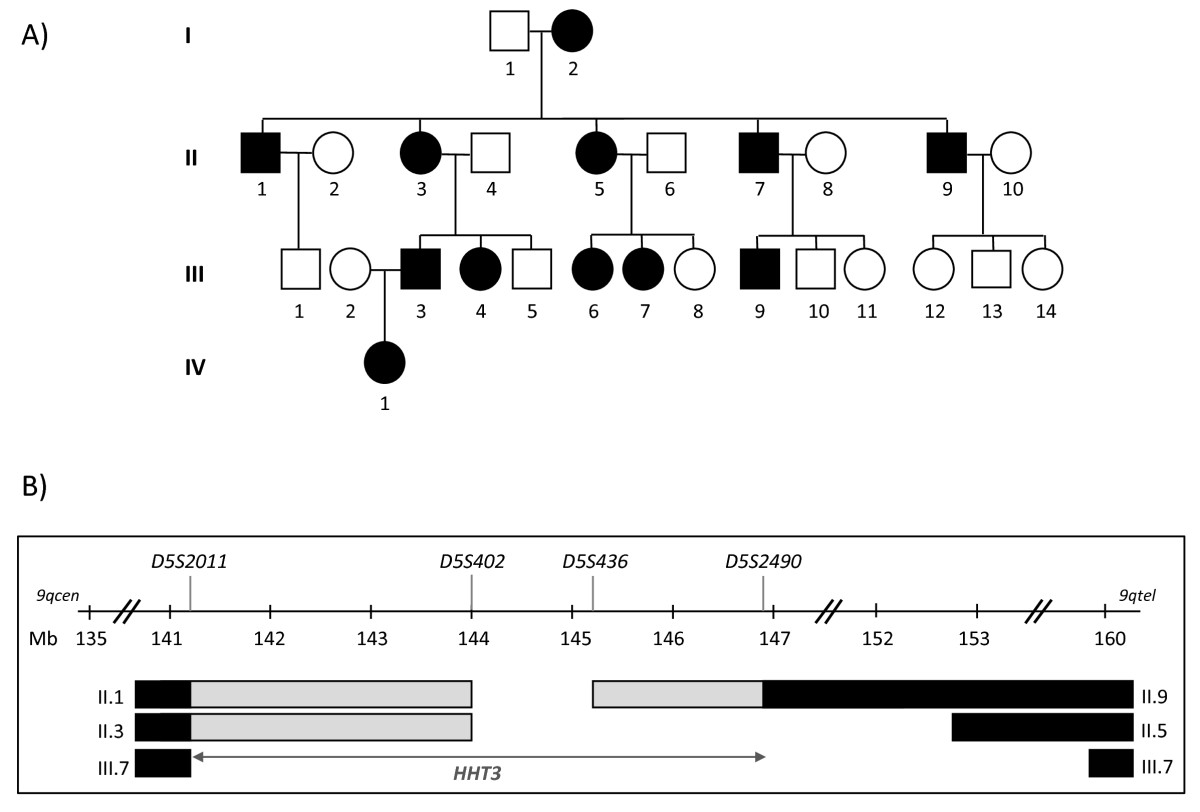
Figure 1
Figure 1 caption
Previousinterval. A) linked Family S pedigree (black symbols represent HHT-affected individuals; white symbols represent unaffected individuals; squares represent males and circles represent females. Individuals are denoted by their generation number, such that the oldest male is I.1, oldest female I.2). B) Published interval on chromosome 5 between microsatellite markers and [], identifying the region shared by all affected individuals (open interval between and ); regions where the designated individuals had inherited their affected parent's non disease-gene bearing allele (black bars), and regions in which the transition from the non disease-gene bearing allele (black bars) to disease-gene bearing allele (open interval) had occurred, but the exact sites of the meiotic recombination event was not definable using the markers studied in [] (grey bars; uninformative markers). It was possible that the interval could be reduced to a minimum of , according to where the recombination breakpoints had occurred in individuals II.1, II.3 and II.9. cen: centromere; tel: telomere; Mb: mega bases.
The published chromosome 5
Excluding any of these genes from the
In this study we report the fine mapping of the published
Methods
Pedigree
Ethics approval was obtained from the Multicentre Research Ethics Committee for Scotland (MREC/98/0/42; 07/MRE00/19), by the Hammersmith, Queen Charlotte's, Chelsea, and Acton Hospital Research Ethics Committee (LREC 99/5637M) and in 2007 by the Hammersmith Hospitals Trust (SHOV1022, 2007). The study is registered on http://www.clinicaltrials.gov (NCT00230620). The pedigree (Family S, Figure 1A) and DNA extractions were as described in [35].
PCR and sequencing
Oligonucleotide primers were ordered from Eurofins MWG-Biotech and amplified in informative members of Family S by polymerase chain reaction (PCR). Genomic primers spanning exons and > 30 bp of flanking intronic sequences were amplified by PCR using
Fine mapping
Non-disease causing genomic sequence variants found during
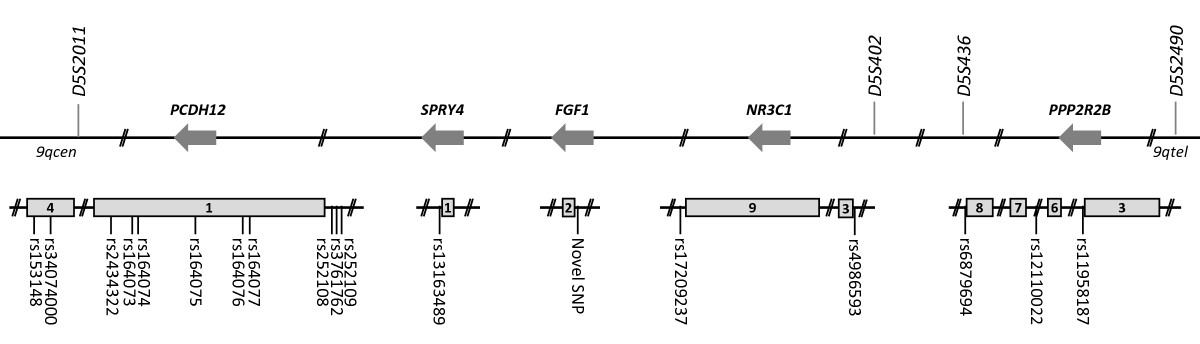
Figure 2
Figure 2 caption
Informative genomic sequence variants used in fine mapping. Locations of genomic variants where individual I.2 was heterozygous, potentially allowing her disease associated and non-disease associated alleles to be tracked. Sequence variants are illustrated in relation to candidate gene exons (grey boxes) and original microsatellite markers. By chance, all five genes are on the reverse strand of chromosome 5, designated by reverse arrows. as on NCBI dbSNP.

Figure 3
Figure 3 caption
Genomic variant haplotypes in interval-defining members of the pedigree. A) Allele inheritance and haplotypes for the inheritance of the key microsatellite markers, and new genomic sequence variants, across and genes in eight members of Family S. For each sequence variant, offspring inherit one allele from their mother and one from their father. Occasional alleles where it was not possible to determine parental origin (and which were therefore not used for haplotype construction) are denoted in non-bold italics. Individual II.5 demonstrates the disease haplotype across the interval, a haplotype which is now shared by II.1, but not II.3 and her descendants until in . Therefore II.3 excludes the centromeric region between and the novel SNP in . B) Haplotypes for the inheritance of the new genomic sequence variants across the gene in nine members of Family S. Affected individuals II.1, II.3, II.5, III.3 and III.4 demonstrate the disease haplotype which is not shared by II.9 at , but is shared at , and the remainder of the interval. Therefore II.9 excludes and beyond from the telomeric extreme of the interval.]: uninformative regions; bp: base pairs.
For confirmations of genotypes, SNPs
Genome analysis
Chromosome 5 positions of genomic sequence variants are based on NCBI Reference build 36 (hg18) assembly.
Results
The
Figure 3 tracks the inheritance of alleles at these SNPs and triplet repeat in the centromeric (Figure 3A), and telomeric (Figure 3B) extremes of the interval. Although I.2 shared three genotypes with I.1, it was possible to definitely state which of I.2's alleles had been inherited by each of their children at all sites except
As shown in Figure 3A, sequencing 15 of these markers allowed better definition of the centromeric regions where II.1 and II.3 had inherited the disease-gene associated allele, and indicated that the site of the recombination breakpoint differed in the two individuals. In individual II.1, the breakpoint had occurred close to
These data in individual II.3 indicated that in addition to the definite
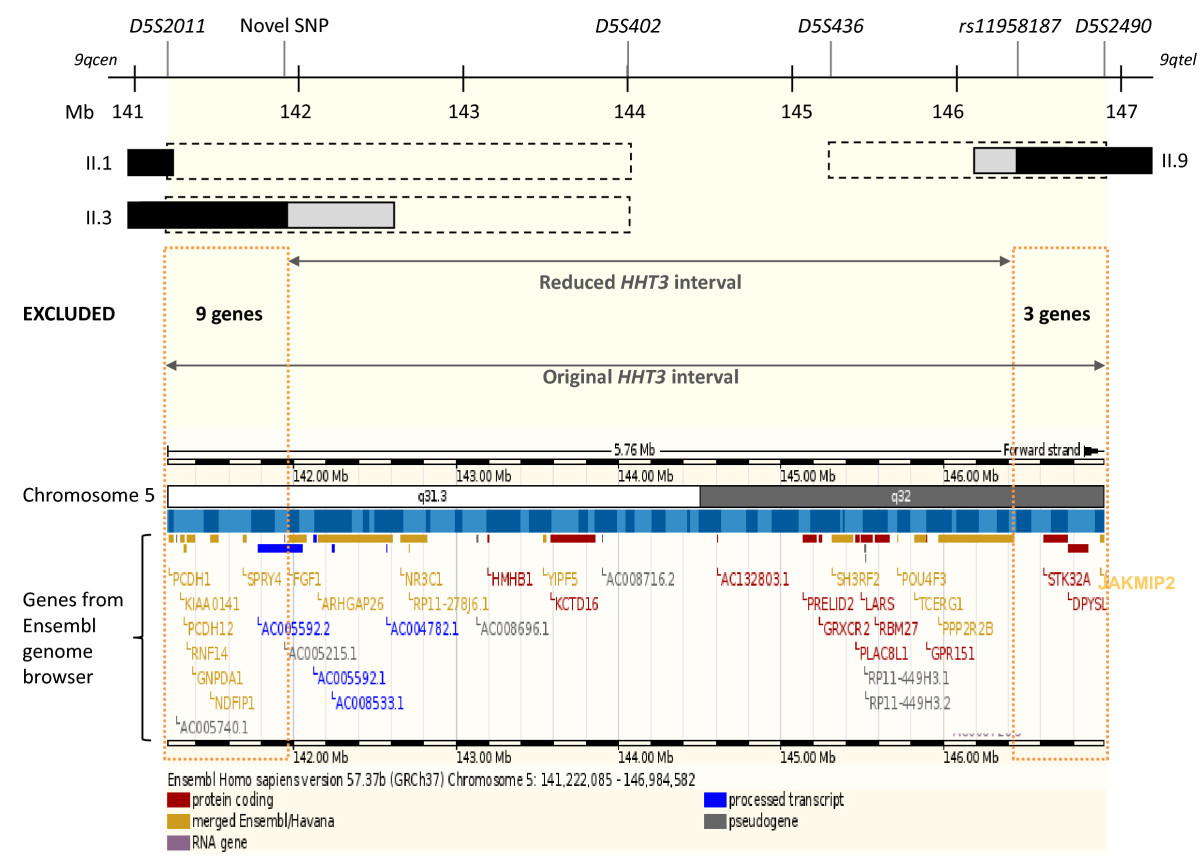
Figure 4
Figure 4 caption
The refinedinterval. The original interval was between microsatellite markers and , and uninformative between and (black dotted bars) in key individuals II.1, II.3 and II.9 []. Informative genomic variations used in fine mapping reduced the extent of the uninformative regions (grey bars, see Figure 1B). The refined interval is sited between a novel SNP within intron 1, and within intron 4. Black bars indicate the definite recombination events that defined the interval in the denoted individuals. Fine mapping, by reducing the interval, excluded 12 interval genes as shown in the lower Ensembl genome browser image of candidate genes (genes no longer in the interval are denoted by orange dotted boxes; Mb: mega bases).
The telomeric border of the
Discussion
Identification of further HHT genes is predicted to provide new insights into TGF-β/BMP signalling and angiogenesis pathways. The previously published
It was recognised that fine mapping of the genomic variants found during candidate gene sequencing could help define the critical recombination events and exclude certain genes, including those for VE-cadherin-2 and Sprouty4. Hence candidate gene sequence variants were examined in the interval-defining members of the Family S pedigree that included the crucial individuals whose recombination events had defined the centromeric and telomeric borders of the
To optimise PCR fidelity, candidate genes and genomic sequence variants were amplified with
Confirmed genotypes allowed construction of haplotypes across the centromeric and telomeric extremes of the
The original interval contained 38 genes according to the Ensembl genome browser. Twelve have been excluded in this study. The 26 genes that remain include 10 pseudogenes or processed transcripts with no known protein products; two genes of potential functional relevance to angiogenesis or TGF-β signalling (
Conclusions
Further examination of key recombination events in a known
Acknowledgements
This work was funded by the British Heart Foundation. We are also grateful for support from the NIHR Biomedical Research Centre Funding Scheme. The study sponsors played no part in study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. The authors thank Dr Gillian Wallace and Dr Stewart Cole for DNA extractions; Dr Mick Jones and Dr Carol Shoulders for helpful discussions; and the family for their willing participation in these studies.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2
Authors’ original file for figure 3
Authors’ original file for figure 4
References
- The logic of TGFbeta signaling. FEBS Lett. 2006;580(12):2811-2820.
- Specificity, diversity, and regulation in TGF-beta superfamily signaling. FASEB J. 1999;13(15):2105-2124.
- Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol. 2008;28(22):6889-6902.
- ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111(2):633-642.
- Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J Biol Chem. 2009;284(23):15794-15804.
- Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12(4):817-828.
- Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA. 2000;97(6):2626-2631.
- Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem. 2005;280(30):27800-27808.
- Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17(7):860-871.
- Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8(4):345-351.
- Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13(2):189-195.
- A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). The Lancet. 2004;363(9412):852-859.
- Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11(1):79-89.
- Emerging role of bone morphogenetic proteins in angiogenesis. Cytokine Growth Factor Rev. 2009;20(3):203-212.
- Diagnostic Criteria for Hereditary Hemorrhagic Telangiectasia (Rendu-Osler-Weber Syndrome). American Journal of Medical Genetics. 2000;91(1):66-67.
- A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104(10):1343-1351.
- Defective angiogenesis in mice lacking endoglin. Science. 1999;284(5419):1534-1537.
- Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol. 2000;217(1):42-53.
- Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet. 2000;26(3):328-331.
- A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Hum Mol Genet. 2003;12(5):473-482.
- Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development. 2002;129(12):3009-3019.
- Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119(11):3487-3496.
- Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res. 2010;106(8):1425-1433.
- Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transpl. 2008;14(2):210-213.
- Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. 2010;16(4):420-428.
- Hereditary haemorrhagic telangiectasia: Pathogenesis, diagnosis and treatment. Blood Reviews. 2010.
- Inhibition of endothelial cell movement by pericytes and smooth muscle cells: activation of a latent transforming growth factor-beta 1-like molecule by plasmin during co-culture. J Cell Biol. 1989;109(1):309-315.
- Endoglin plays distinct roles in vascular smooth muscle cell recruitment and regulation of arteriovenous identity during angiogenesis. Dev Dyn. 2009;238(10):2479-2493.
- Compensatory signalling induced in the yolk sac vasculature by deletion of TGFbeta receptors in mice. J Cell Sci. 2007;120(Pt 24):4269-4277.
- Reduced endothelial secretion and plasma levels of transforming growth factor-beta1 in patients with hereditary hemorrhagic telangiectasia type 1. Cardiovasc Res. 2005;68(1):155-164.
- Update on molecular diagnosis of hereditary hemorrhagic telangiectasia. Hum Genet. 2010;128(1):61-77.
- Likelihood ratios to assess genetic evidence for clinical significance of uncertain variants: hereditary hemorrhagic telangiectasia as a model. Exp Mol Pathol. 2008;85(1):45-49.
- Multiple sequence variants in hereditary hemorrhagic telangiectasia cases: illustration of complexity in molecular diagnostic interpretation. J Mol Diagn. 2009;11(6):569-575.
- Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am J Med Genet A. 2010;152A(2):333-339.
- A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. Journal of Medical Genetics. 2005;42:577-582.
- A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140(20):2155-2162.
- FGF-1 and S100A13 possibly contribute to angiogenesis in endometriosis. J Reprod Immunol. 2005;67(1-2):87-101.
- Structure of the 55-kDa regulatory subunit of protein phosphatase 2A: evidence for a neuronal-specific isoform. Biochemistry. 1991;30(15):3589-3597.
- The protein kinase complement of the human genome. Science. 2002;298(5600):1912-1934.
- cDNA cloning, chromosomal mapping, and expression analysis of human VE-Cadherin-2. Mamm Genome. 2000;11(11):1030-1033.
- Gene expression fingerprinting for human hereditary hemorrhagic telangiectasia. Hum Mol Genet. 2007;16(13):1515-1533.
- Angiogenesis: the VE-cadherin switch. Trends Cardiovasc Med. 2006;16(2):55-59.
- VE-cadherin is a critical endothelial regulator of TGF-beta signalling. Embo J. 2008;27(7):993-1004.
- Sprouty proteins: antagonists of endothelial cell signaling and more. Thromb Haemost. 2003;90(4):586-590.
- Inhibition of angiogenesis by a mouse sprouty protein. J Biol Chem. 2001;276(6):4128-4133.
- Sprouty2 and Sprouty4 are essential for embryonic morphogenesis and regulation of FGF signaling. Biochem Biophys Res Commun. 2007;352(4):896-902.
- Sprouty4 deficiency potentiates Ras-independent angiogenic signals and tumor growth. Cancer Sci. 2009;100(9):1648-1654.
- One-step enzymatic purification of PCR products for direct sequencing. Curr Protoc Hum Genet. 2001.
- A rapid, inexpensive method for eluting DNA from agarose or acrylamide gel slices without using toxic or chaotropic materials. Biotechniques. 1992;13(2):205-206.
- Purification and concentration of DNA from aqueous solutions. Curr Protoc Immunol. 2001.
- Variations of the human glucocorticoid receptor gene (NR3C1): pathological and in vitro mutations and polymorphisms. Hum Mutat. 2003;21(6):557-568.
- Enhancement of PCR amplification yield and specificity using AmpliTaq Gold DNA polymerase. Biotechniques. 1998;25(4):716-722.
- Phusion™ Hot Start High-Fidelity DNA Polymerase (manual). [http://www.neb.com/nebecomm/ManualFiles/manualF-540.pdf]
- Prevention of pre-PCR mis-priming and primer dimerization improves low-copy-number amplifications. Nucleic Acids Res. 1992;20(7):1717-1723.
- Regulation of endothelial cell gene expression: insights from HHT3 candidate gene studies. PhD thesis. 2009.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.