Autocrine activity of soluble Flt-1 controls endothelial cell function and angiogenesis
Vascular Cell. 2011;
Received: 4 April 2011 | Accepted: 13 July 2011 | Published: 13 July 2011
Vascular Cell ISSN: 2045-824X
Abstract
Background
The negative feedback system is an important physiological regulatory mechanism controlling angiogenesis. Soluble vascular endothelial growth factor (VEGF) receptor-1 (sFlt-1), acts as a potent endogenous soluble inhibitor of VEGF- and placenta growth factor (PlGF)-mediated biological function and can also form dominant-negative complexes with competent full-length VEGF receptors.
Methods and results
Systemic overexpression of VEGF-A in mice resulted in significantly elevated circulating sFlt-1. In addition, stimulation of human umbilical vein endothelial cells (HUVEC) with VEGF-A, induced a five-fold increase in sFlt-1 mRNA, a time-dependent significant increase in the release of sFlt-1 into the culture medium and activation of the
Conclusion
These results show that endothelial sFlt-1 expression is regulated by VEGF and acts as an autocrine regulator of endothelial cell function.
Background
Vascular endothelial growth factor-A (VEGF-A) is a multifunctional cytokine induced by hypoxic stress [1]. It plays a pivotal role in many aspects of embryonic cardiovascular development, including formation of blood vessels, cardiac morphogenesis, and development of the nervous system [2–6]. Loss and gain of function studies in mice indicate that VEGF-A levels have to be maintained within a narrow range to ensure proper cardiovascular development and embryo survival [7–9]. It has been shown that the effects of VEGF-A can be deleterious if uncontrolled. Over-expression of VEGF in experimental animals increases the leakiness of blood vessels, which may lead to severe edema, loss of limb and death [10, 11]. Excess VEGF-A expression in skeletal muscle results in the induction of vascular tumors (hemangiomas) [12–14], whereas loss of VEGF-A activity due to increased production of its natural antagonist, sFlt-1 (soluble VEGF receptor-1/sVEGFR-1), as in preeclampsia, reduces angiogenesis [15]. Thus, homeostasis requires mechanisms to regulate the functional activity of VEGF-A.
Soluble Flt-1 is generated by alternative splicing of the fms-like tyrosine kinase (
Materials and methods
Reagents
Recombinant growth factors were purchased from RELIATech (Brauschweig, Germany). Rabbit polyclonal antibodies against phospho- endothelial nitric oxide synthase (eNOS) at serine-1177 (p-eNOSSer1177), phospho-ERK-1/-2 MAPK and phospho-VEGF receptor-2 (VEGFR-2) tyrosine-951 antibodies were purchased from Calbiochem (Nottingham, UK). Small inhibitory RNAs (siRNA) and oligonucleotide primers were purchased from Eurogentec (Southampton, UK). Luciferase reporter assay and cDNA synthesis kits were from Promega (Southampton, UK). All other cell culture reagents and chemicals were obtained from Sigma Aldrich (Poole, UK).
Placental tissues
Human placental tissue was obtained from normal pregnancies and gestationally-matched pregnancies complicated by preeclampsia. Preeclampsia was defined as blood pressure > 140/90 mm Hg on at least two consecutive measurements and proteinuria of at least 300 mg per 24 hours. Informed consent was obtained from the patients and the study had the approval of the South Birmingham Ethical Committee (Birmingham, UK).
Cell Culture
Primary human umbilical vein endothelial cells (HUVEC) were isolated and cultured as described [26]. Cells were used at passage two or three for experiments and serum-starved in endothelial cell serum-free medium (Gibco-BRL, UK) supplemented with 0.2% bovine serum albumin for 24 hours prior to stimulation.
Adenoviral gene transfer
The recombinant, replication-deficient adenoviruses encoding sFlt-1 (Ad-sFlt-1) VEGF (Ad-VEGF) and PTEN (Ad-PTEN) were used as described previously [27–29].
Quantitative Real-Time PCR
Sample preparation and real-time PCR was performed as described previously [30]. Briefly, mRNA was prepared using TRIzol and DNase-1 digestion/purification on RNAeasy columns (Qiagen), and reverse transcribed with the cDNA Synthesis Kit (Promega). Triplicate cDNA samples and standards were amplified in SensiMix containing SYBR green (Quantace) with primers specific for sFlt-1 [31]. The mean threshold cycle (CT) was normalized to β-actin and expressed relative to control.
siRNA knock-down of sFlt-1
Two siRNA sequences to the unique 3' sequence of sFlt-1 (sFlt-1 A
Transduction of chimeric VEGF Receptors in HUVEC
A chimeric VEGF/epidermal growth factor (EGF) receptor comprising the intracellular and transmembrane domains of VEGFR-2 fused to the extracellular domain of the human EGF receptor [33]. EGF does not bind to VEGF receptors, therefore, it does not activate the endogenous VEGF receptors. EGDR and its tyrosine-to-phenylalanine mutants (EGDR-Y951F) were generated and cloned into the pMMP retroviral vector, and retrovirus-containing cell supernatant was harvested and used immediately to infect HUVEC [33]. Following 16 hours of incubation, the medium was replaced with fresh growth medium and the HUVEC were used 48 hours after infection.
Nitric oxide (NO) Release
Total NO in conditioned media was assayed as nitrite, the stable breakdown product of NO, using a Sievers NO chemiluminescence analyzer (Analytix, Sunderland, UK) as described previously [33].
Tube Formation Assay
The formation of capillary-like structures was examined on growth factor-reduced Matrigel in 24-well plates as described previously [33]. Tube formation was quantified by measuring the total tube length in five random x200 power fields per well using a Nikon phase-contrast inverted microscope with Image ProPlus image analysis software (Media Cybernetics, Silver Spring, USA). Mean total tube length was calculated from three independent experiments performed in duplicate.
flt-1gene promoter activity assay
A 1.3 Kb fragment of the human
Western Blotting
Cells lysates were immunoblotted as described previously [33]. Membranes were probed with rabbit polyclonal antibodies against phospho-eNOS-Ser1177, anti-ERK-1/-2 or anti-VEGFR-2 phosphotyrosine-951 at 4°C overnight. Proteins were visualised using the ECL detection kit (Amersham-Pharmacia, UK).
sFlt-1 ELISA
Soluble Flt-1 (sFlt-1) levels in culture supernatants were measured as previously described [30].
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissues were used for immunohistochemistry as previously described [15].
Statistical analysis
All data are expressed as mean ± SEM. Statistical comparisons were performed using one-way ANOVA followed by the Student-Newman-Keuls test as appropriate. Statistical significance was set at a value of p < 0.05.
Results and Discussion
VEGF-A stimulates sFlt-1 release
To evaluate the capacity of VEGF-A to regulate the secretion of its negative regulator, sFlt-1, HUVEC were incubated with VEGF-A and the conditioned media assayed for sFlt-1 by ELISA. VEGF-A stimulated a concentration and time dependent increase in the release of sFlt-1 from HUVEC that reached a maximum at 20 ng/ml and 48 hours, respectively (Figure 1a and 1b). Consistent with these findings, qPCR revealed a greater than five-fold increase in sFlt-1 mRNA after 22 hours of VEGF-A stimulation (Figure 1c). In addition, VEGF-A induced
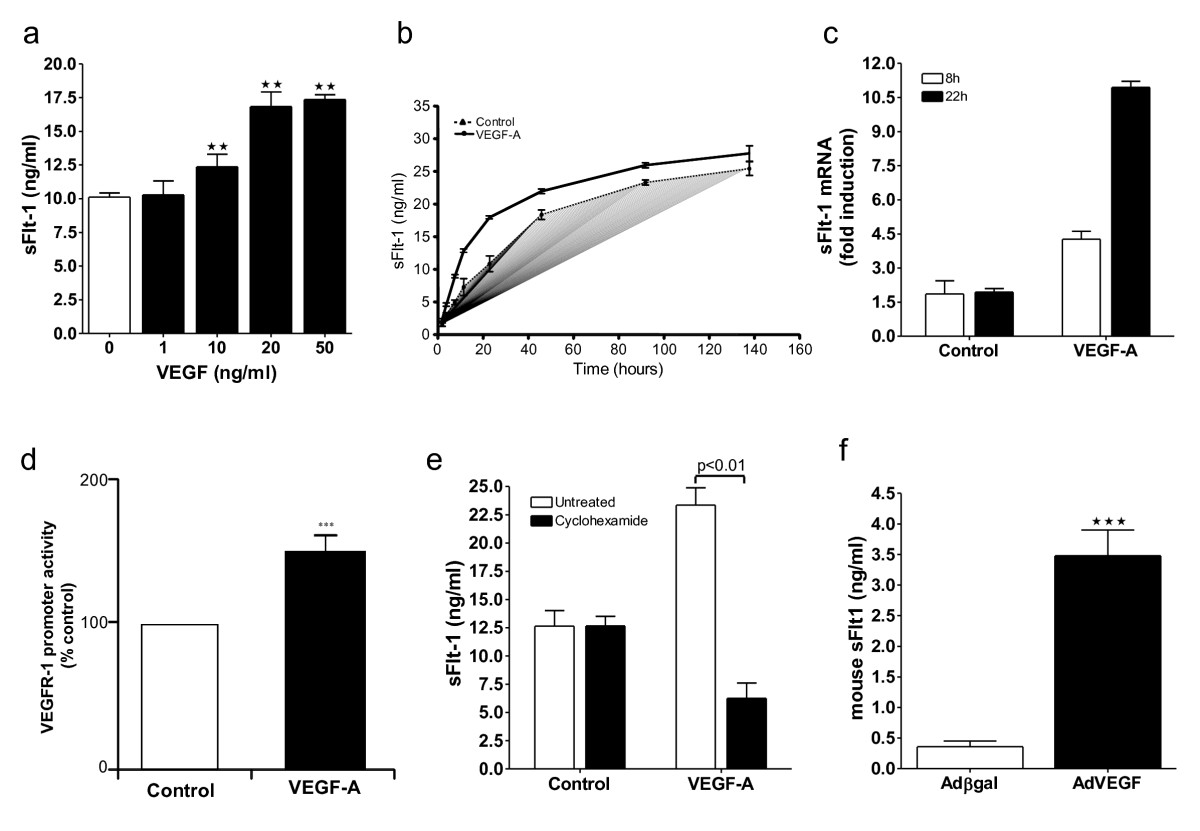
Figure 1
Figure 1 caption
VEGF stimulates sFlt-1 release from endothelial cells in vitro, and in vivo. (a) VEGF stimulated sFlt-1 release from HUVEC in a concentration-dependent manner. sFlt-1 was measured, in the culture medium, by ELISA, following a 24-hour incubation. (b) Time course of VEGF (20 ng/ml) induced sFlt-1 release from HUVEC. (c) VEGF induced an increase in sFlt-1 mRNA levels in HUVEC as determined by qPCR and, (d) promoter activity in porcine aortic endothelial cells by luciferase assay. (e) Cyclohexamide (10 μg/ml) inhibits VEGF-induced sFlt-1 protein synthesis after 24 hours of treatment. (f) Mouse plasma levels of sFlt-1 five days post intravenous injection with an adenovirus encoding VEGF (Ad-VEGF) or control (Ad-βgal). Data are expressed as mean (± SEM) or representative of three or more independent experiments performed in triplicate. ** < 0.01; *** < 0.001 vs. control.
Activation of VEGFR-2, mediates the release of sFlt-1
To identify the VEGF receptors involved in the release of sFlt-1, HUVEC were stimulated with either VEGF-A (binds VEGFR-1 and VEGFR-2), or PlGF-1 (binds VEGFR-1) or VEGF-E (binds VEGFR-2). PlGF-1 showed no effect on sFlt-1 release, whereas VEGF-E stimulated similar levels of sFlt-1 release to those induced by VEGF-A (Figure 2a), suggesting that release of sFlt-1 is mediated by VEGFR-2. Preincubation of endothelial cells with SU1498, a VEGFR-2 selective inhibitor, blocked the VEGF-A induced sFlt-1 release (Figure 2b), confirming the importance of this receptor for the response.
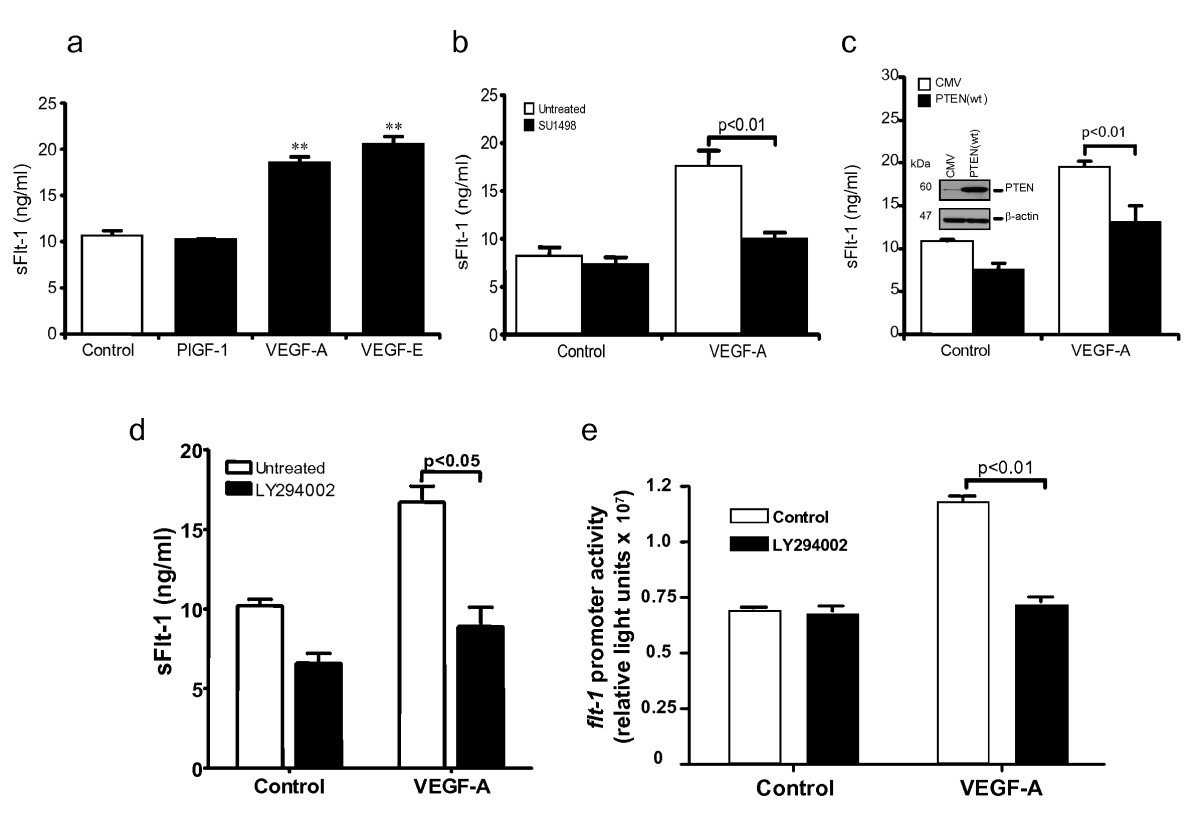
Figure 2
Figure 2 caption
VEGF-A-induced sFlt-1 release requires VEGFR-2 and the PI3K pathway. (a) sFlt-1 release from HUVEC 24 hours after incubation with PlGF-1 (20 ng/ml), VEGF-A (20 ng/ml) and VEGF-E (20 ng/ml). (b) sFlt-1 release from HUVEC after incubation with SU1498 (10 μM), a VEGFR-2 tyrosine kinase inhibitor, prior to addition of VEGF-A (20 ng/ml) for 24 hours. (c) sFlt-1 release from HUVEC infected with an adenovirus encoding PTEN (PTEN (wt)) or control adenovirus (CMV) prior to stimulation with VEGF-A (20 ng/ml) for 24 hours. Western blot demonstrating decreased PTEN expression in HUVEC infected with Ad-PTEN(wt) compared with Ad-CMV control (insert). (d) sFlt-1 release from HUVEC after incubation with LY294002 (10 μM), a PI3K inhibitor, prior to addition of VEGF-A (20 ng/ml) for 24 hours. (e) LY294002 prevented the VEGF-A-induced promoter activity in porcine aortic endothelial cells, assayed by luciferase reporter assay. Data are expressed as mean (± SEM) or representative of three or more independent experiments performed in triplicate. ** < 0.01 vs. control.
VEGF stimulated sFlt-1 production, is mediated via PI3K
To investigate the role of the PI3K pathway in VEGF-A-induced sFlt-1 release, PI3K activity was inhibited through overexpression of PTEN (Phosphatase and Tensin homolog deleted on chromosome Ten), which dephosphorylates phosphatidylinositol 3,4,5-triphosphate and has been shown to reduce VEGF-mediated signaling and cellular function [28, 35]. HUVEC were infected overnight with an adenovirus encoding PTEN (PTEN(wt)) or a control adenovirus (CMV) and stimulated with VEGF-A for 24 hours. Inhibition of PI3K activity by PTEN overexpression led to a significant decrease in sFlt-1 release (Figure 2c). Furthermore, pre-incubation of HUVEC with LY294002, a pharmacological PI3K inhibitor, also attenuated the VEGF mediated release of sFlt-1 (Figure 2d) and of
Loss of sFlt-1 promotes angiogenesis
Adenoviral-mediated overexpression of sFlt-1 in HUVEC inhibited endothelial cell proliferation (Figure 3a) and MAP kinase ERK-1/-2 phosphorylation (Figure 3a insert). Subsequently, to test whether knockdown of sFlt-1 would promote endothelial cell proliferation, HUVEC were transfected with two synthetic siRNA sequences targeted to the unique carboxyl-terminus region of sFlt-1. sFlt-1 siRNA transfection resulted in a substantial reduction in the release of sFlt-1 from HUVEC after 24 hours (Figure 3b). Endothelial cell proliferation was significantly increased (Figure 3c) and interestingly, sFlt-1 knockdown also led to a concomitant increase in VEGFR-2 phosphorylation at tyrosine 951 (Y951) (Figure 3d). In addition, sFlt-1 siRNA increased both basal and VEGF-A-mediated endothelial cell migration (Figure 4a) and tube formation on Matrigel (Figure 4b and 4c). VEGF stimulates eNOS activity and NO release [23, 36] to mediate angiogenesis [33, 37], thus we predicted that loss of sFlt-1 would increase eNOS phosphorylation in HUVEC. Phosphorylation of eNOS (ser1177) was significantly increased in cells lacking sFlt-1 (Figure 4d). These data provide direct evidence that sFlt-1 is itself a negative regulator of endothelial function. It is likely that sFlt-1 sequesters VEGF and PlGF to maintain a physiological steady state until angiogenesis is required, at which point this system must be overridden.
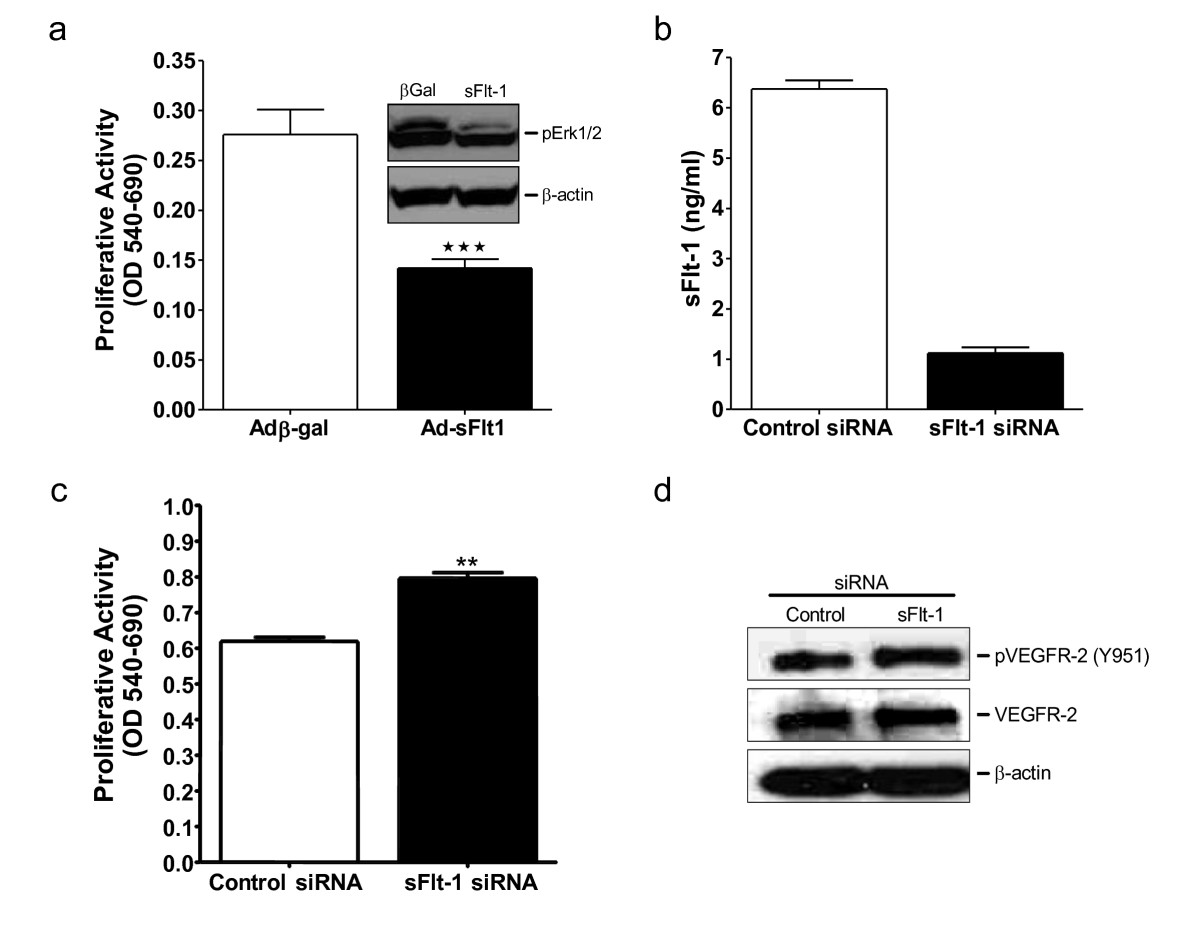
Figure 3
Figure 3 caption
sFlt-1 inhibits endothelial cell proliferation and VEGF receptor phosphorylation. (a) Cell proliferation after 48 hours of treatment in HUVEC infected with an adenovirus encoding sFlt-1 (Ad-sFlt1) or β-galactosidase control (Ad-βGal). Western blot demonstrating decreased Erk1/2 phosphorylation in HUVEC infected with Ad-sFlt-1 compared with Ad-β-gal control (insert). (b) Dramatic reduction of sFlt-1 release from HUVEC 24 hours after transfection with sFlt-1 siRNA. (c) Cell proliferation after 48 hours in sFlt-1 siRNA transfected HUVEC. (d) Western blot showing VEGF receptor-2 (VEGFR-2) phosphorylation at tyrosine 951 in HUVEC transfected with sFlt-1, or control siRNA. VEGFR-2 and β-actin were used as a loading control. Data are expressed as mean (± SEM) or representative of three or more independent experiments performed in triplicate. ** < 0.01 vs. control.
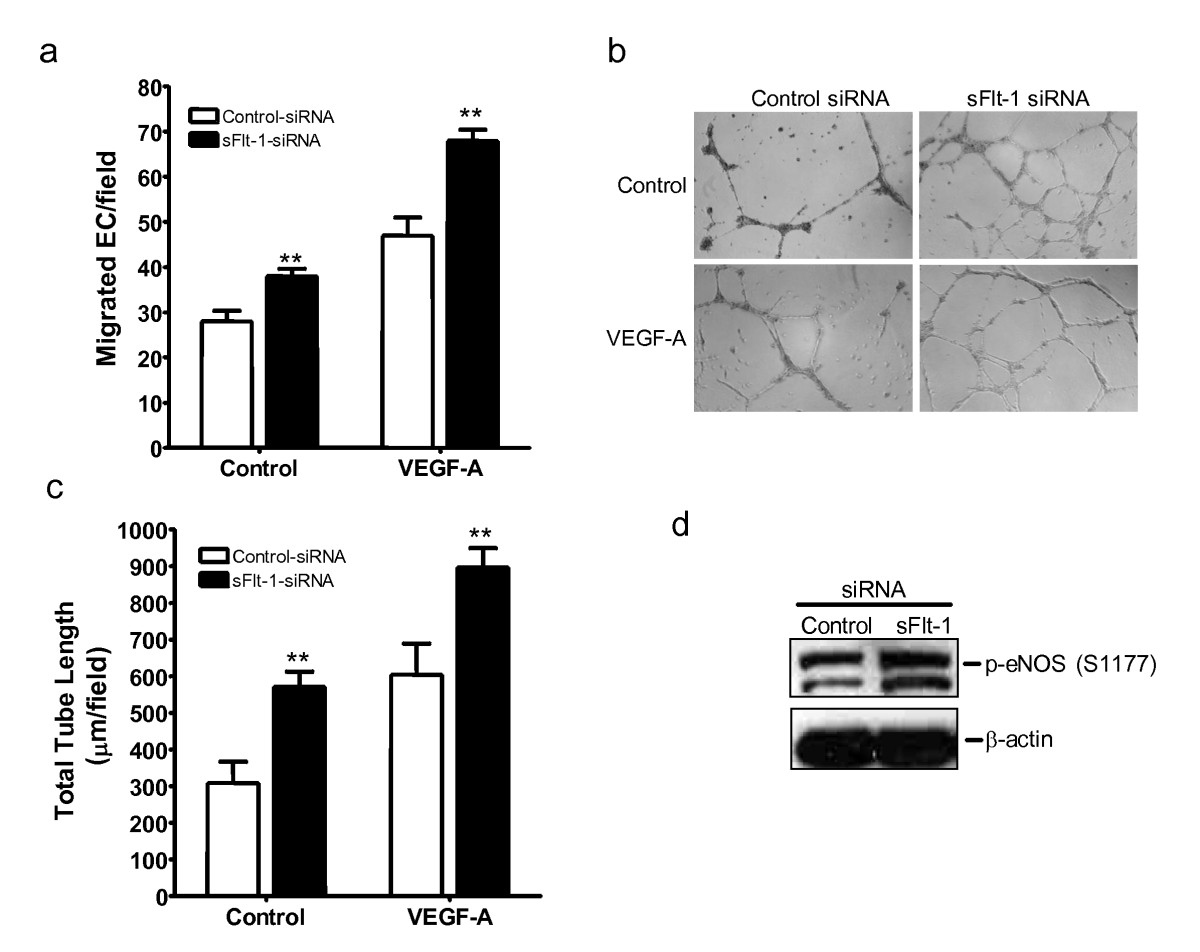
Figure 4
Figure 4 caption
Loss of sFlt-1 enhances angiogenesis and eNOS phoshorylation. (a) VEGF (20 ng/ml) induced an increase in cell migration of HUVEC transfected with sFlt-1 siRNA compared with control siRNA using a modified Boyden chamber assay. (b) VEGF-induced capillary-like tube formation and (c) quantification of mean total tube length per field of view after six hours treatment in HUVEC transfected with sFlt-1 siRNA or control. (d) Representative Western blot showing eNOS phosphorylation at serine 1177 (p-eNOS (S1177)) in HUVEC transfected with soluble (sFlt-1) or control siRNA. β-actin was used as a loading control. Data are expressed as mean (± SEM) or representative of three or more independent experiments performed in triplicate. ** < 0.01 vs. control.
Excess sFlt-1 inhibits VEGFR-2 Y951 phosphorylation
Activation of VEGFR-2 leads to an increase in eNOS expression and activation,[38] which is essential for neovascularisation [37]. A recent study showed that mutation of VEGFR-2 Y951 to phenylalanine caused a significant reduction in VEGF-induced angiogenesis [33]. As preeclampsia is associated with elevated placental [15] and circulating [22] sFlt-1 and placental sFlt-1 inhibits angiogenesis [15], we speculated that elevated free sFlt-1 would lead to a reduction of VEGFR-2 phosphorylation in preeclamptic placenta. Using the EGFR-chimeric receptor system we show that mutation of Y951 to phenylalanine resulted in over 50% reduction in NO release (Figure 5a) and overexpression of sFlt-1 in endothelial cells abrogated phosphorylation of VEGFR-2 Y951 (Figure 5b). To assess whether VEGFR-2 phosphorylation was reduced in preeclamptic placenta, that express elevated sFlt-1, we undertook immunohistochemical analysis for phospho-VEGFR-2 Y951. Overall, phosphorylation of VEGFR-2 Y951 was greatly reduced in the preeclamptic placenta compared to gestationally-matched, normal placenta (Figure 5c). Together, these findings indicate that increased levels of sFlt-1 have a negative effect on VEGFR-2 tyrosine phosphorylation, which in turn would lead to concomitant inhibition of downstream function and signaling and compromise maternal vascular homeostasis and placental angiogenesis.
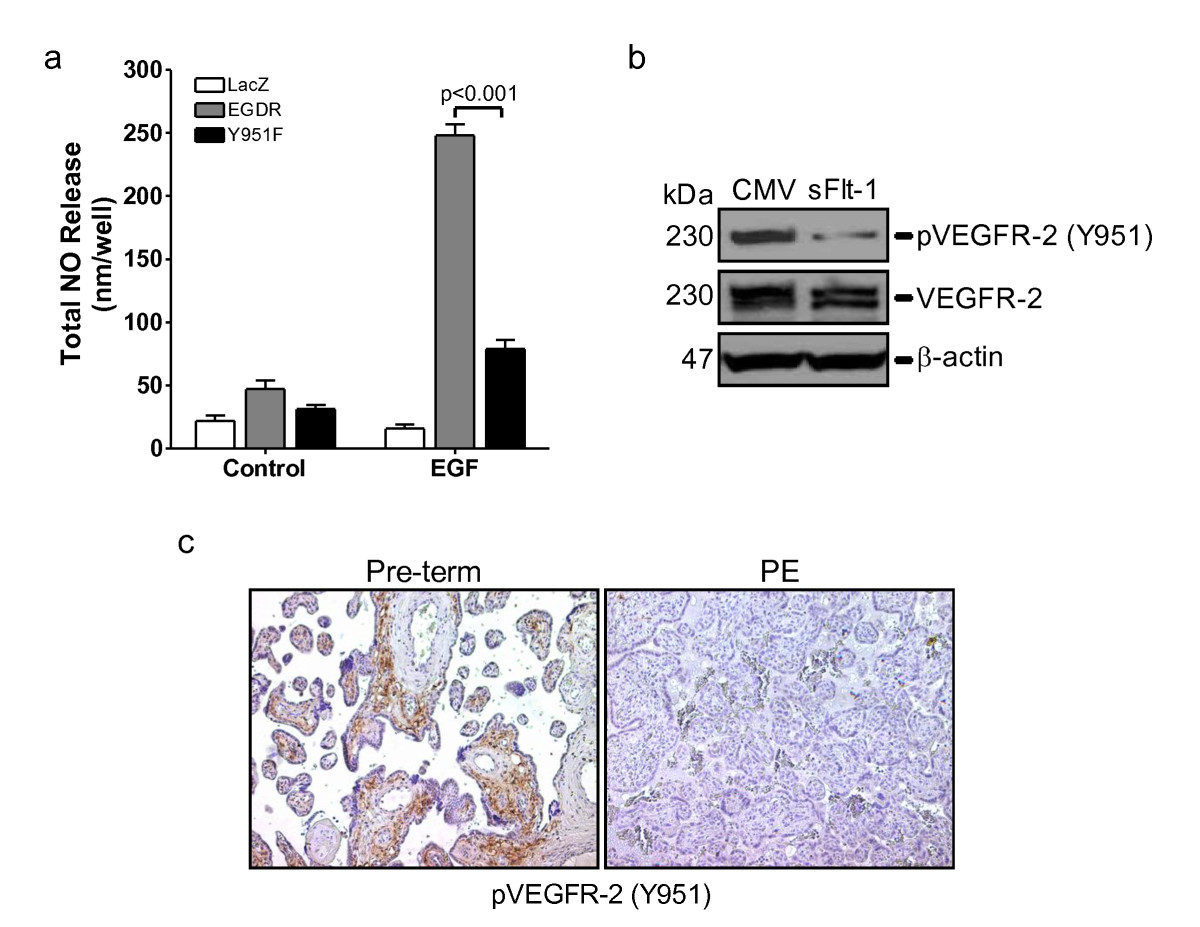
Figure 5
Figure 5 caption
Excess sFlt-1 inhibits VEGF receptor phosphorylation at tyrosine 951. (a) Nitric oxide (NO) release from HUVEC overexpressing chimeric EGF receptor/VEGFR-2 constructs (EGDR) and EGDR with a mutated Y951 (Y951F), following stimulation with EGF (10 ng/ml) for one hour. NO was measured in cell supernatants using a Sievers chemiluminescent NO analyzer and background subtracted. (b) Western blot showing VEGFR-2 and VEGF-2 phosphorylation at tyrosine 951 (p-VEGFR-2 (Y951)) in HUVEC infected with an adenovirus encoding sFlt-1 (sFlt1) or control virus (CMV). β-actin was used as a loading control. (c) Representative immunohistochemical staining for p-VEGFR-2 (Y951) in gestationally-matched normal (pre-term) and preeclamptic (PE) placenta. Data are expressed as mean (± SEM) or representative of three or more independent experiments performed in triplicate.
Conclusions
Endothelial cell sFlt-1 expression is regulated by VEGF and sFlt-1 is an autocrine regulator of endothelial cell function.
Acknowledgements and funding
This work was supported by grants from the Medical Research Council (G0600270, G0601295 and G0700288) and British Heart Foundation (RG/09/001/25940 and PG/06/114).
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2
Authors’ original file for figure 3
Authors’ original file for figure 4
Authors’ original file for figure 5
References
- Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843-845.
- A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128:1531-1538.
- The biology of VEGF and its receptors. Nat Med. 2003;9:669-676.
- A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA. 2001;98:5780-5785.
- Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131-138.
- Treatment of motoneuron degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS. Nat Neurosci. 2005;8:85-92.
- Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435-439.
- Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439-442.
- Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941-3946.
- Angiogenic gene therapy for experimental critical limb ischemia: acceleration of limb loss by overexpression of vascular endothelial growth factor 165 but not of fibroblast growth factor-2. Circ Res. 2002;90:966-973.
- Evaluation of angiogenesis and side effects in ischemic rabbit hindlimbs after intramuscular injection of adenoviral vectors encoding VEGF and LacZ. J Gene Med. 2002;4:371-380.
- VEGF gene therapy: stimulating angiogenesis or angioma-genesis?. Nat Med. 2000;6:1102-1103.
- VEGF gene delivery to myocardium: deleterious effects of unregulated expression. Circulation. 2000;102:898-901.
- VEGF gene delivery to muscle: potential role for vasculogenesis in adults. Mol Cell. 1998;2:549-558.
- Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circ Res. 2004;95:884-891.
- Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun. 1996;226:324-328.
- Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J Biol Chem. 1994;269:25646-25654.
- Differential binding characteristics and cellular inhibition by soluble VEGF receptors 1 and 2. Exp Cell Res. 1998;241:161-170.
- . Regulation of soluble VEGFR-1 by VEGF and oxygen and its elevation in pre-eclampsia and fetal growth restriction. 2001.
- Vascular endothelial growth factor ligands and receptors that regulate human cytotrophoblast survival are dysregulated in severe preeclampsia and hemolysis, elevated liver enzymes, and low platelets syndrome. Am J Pathol. 2002;160:1405-1423.
- Amniotic fluid--soluble vascular endothelial growth factor receptor-1 in preeclampsia. Obstet Gynecol. 2000;95:353-357.
- Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672-683.
- Role of VEGF receptor-1 (Flt-1) in mediating calcium-dependent nitric oxide release and limiting DNA synthesis in human trophoblast cells. Lab Invest. 1997;76:779-791.
- Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649-658.
- Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691-703.
- Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159:993-1008.
- Adenovirus-mediated delivery of a soluble form of the VEGF receptor Flk1 delays the growth of murine and human pancreatic adenocarcinoma in mice. Surgery. 2002;132:857-865.
- Activation of vascular endothelial growth factor receptor-1 sustains angiogenesis and Bcl-2 expression via the phosphatidylinositol 3-kinase pathway in endothelial cells. Diabetes. 2003;52:2959-2968.
- Reduction of circulating soluble Flt-1 alleviates preeclampsia-like symptoms in a mouse model. J Cell Mol Med. 2010;14:1857-1867.
- Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115:1789-1797.
- Angiotensin II induces soluble fms-Like tyrosine kinase-1 release via calcineurin signaling pathway in pregnancy. Circ Res. 2007;100:88-95.
- Rational siRNA design for RNA interference. Nat Biotechnol. 2004;22:326-330.
- Direct evidence for endothelial vascular endothelial growth factor receptor-1 function in nitric oxide-mediated angiogenesis. Circ Res. 2006;99:715-722.
- Activation of proteinase-activated receptor 2 stimulates soluble vascular endothelial growth factor receptor 1 release via epidermal growth factor receptor transactivation in endothelial cells. Hypertension. 2010;55:689-697.
- PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. J Biol Chem. 2002;277:10760-10766.
- Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131-3139.
- Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest. 1998;101:2567-2578.
- VEGF-A induces expression of eNOS and iNOS in endothelial cells via VEGF receptor-2 (KDR). Biochem Biophys Res Commun. 1998;252:743-746.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.