Notch modulates VEGF action in endothelial cells by inducing Matrix Metalloprotease activity
Vascular Cell. 2011;
Received: 29 December 2010 | Accepted: 18 January 2011 | Published: 18 January 2011
Vascular Cell ISSN: 2045-824X
Abstract
Background
In the vasculature, Notch signaling functions as a downstream effecter of Vascular Endothelial Growth Factor (VEGF) signaling. VEGF regulates sprouting angiogenesis in part by inducing and activating matrix metalloproteases (MMPs). This study sought to determine if VEGF regulation of MMPs was mediated via Notch signaling and to determine how Notch regulation of MMPs influenced endothelial cell morphogenesis.
Methods and Results
We assessed the relationship between VEGF and Notch signaling in cultured human umbilical vein endothelial cells. Overexpression of VEGF-induced Notch4 and the Notch ligand, Dll4, activated Notch signaling, and altered endothelial cell morphology in a fashion similar to that induced by Notch activation. Expression of a secreted Notch antagonist (Notch1 decoy) suppressed VEGF-mediated activation of endothelial Notch signaling and endothelial morphogenesis. We demonstrate that Notch mediates VEGF-induced matrix metalloprotease activity via induction of MMP9 and MT1-MMP expression and activation of MMP2. Introduction of a MMP inhibitor blocked Notch-mediated endothelial morphogenesis. In mice, analysis of VEGF-induced dermal angiogenesis demonstrated that the Notch1 decoy reduced perivascular MMP9 expression.
Conclusions
Taken together, our data demonstrate that Notch signaling can act downstream of VEGF signaling to regulate endothelial cell morphogenesis via induction and activation of specific MMPs. In a murine model of VEGF-induced dermal angiogenesis, Notch inhibition led to reduced MMP9 expression.
Background
Angiogenesis, the formation of new blood vessels from existing vasculature, is a multi-step process that plays a central role in embryogenesis and pathological phenomena. Vascular Endothelial Growth Factor (VEGF) is a key regulator of angiogenesis and is important for the degradation of extracellular matrix (ECM), as well as the subsequent proliferation, migration, and survival of endothelial cells. ECM components, including fibrins, collagens, and laminins, form a lamina around existing vasculature that must be degraded in order to form new vessels.
VEGF signaling via VEGFR-2 induces the expression of endothelial cell-derived matrix metalloproteases (MMPs), including MMP2 [1], MMP9 [2], and MT1-MMP [3], which degrade the matrix to allow for endothelial sprouting. MMPs are thus essential for angiogenesis, and their loss from either endothelium or inflammatory cells has been associated with severe angiogenic defects. At the same time, increased MMP activity has significant vascular consequences. MMPs are antagonized by Tissue Inhibitors of Matrix Metalloproteases (TIMPs), and a pathological increase in endothelial MMP over TIMP activity has been proposed to contribute to vessel wall thickening, abdominal aortic aneurysm formation, varicose veins, hypertension and preeclampsia [4–6]. MMPs are not, however, strictly proangiogenic; other more recently elucidated functions of this protein family include the mediation of vascular regression, as well as the generation of ECM fragments with antiangiogenic properties.
MMP2 and MMP9 belong to the class of gelatinases with collagen and fibrin as substrates and are expressed as inactive pro-proteins. MMP2 and MMP9 are processed to activate their metalloprotease activity. MT1-MMP is a member of the membrane associated or membrane-type MMPs [7–9]. In the vasculature, MT-MMPs are thought to be involved in the localized pericellular release of bioactive growth factors such as bFGF, VEGF, and TGF-beta, processing of pro-MMP2 into an activated MMP2, and degradation of ECM components including collagens and fibrins [10, 11]. MT1-MMP has been shown to be the most potent fibrinolytic MMP and has a critical role in the creation of channels in the ECM, into which endothelial cells migrate during sprouting angiogenesis [12, 13]. MT1-MMP function is also required for proper lumenization of the new vessel [13].
Notch signaling is an evolutionarily conserved pathway that regulates cell fate decisions. Notch proteins, Notch 1 through 4, act as receptors and their ligands Jagged (Jagged-1, 2) and Delta-like (Dll-1, 3, 4), are all transmembrane proteins [14]. Upon ligand binding, the cytoplasmic domain of Notch is released by proteolytic cleavage via presenilin/γ-secretase [15], translocates to the nucleus, and interacts with the transcriptional repressor CSL (CBF1/Su(H)/Lag2), converting it to a transcriptional activator [16]. An essential role for Notch signaling in arterial differentiation and vascular remodeling has been demonstrated by genetic studies of mice with targeted mutations in either Notch (
Signaling via both VEGF and Notch is indispensable in vascular development, and it has become evident that these two pathways are interconnected. Heterozygous deficiency of
In this study, we demonstrate that Notch mediates VEGF-induced MMP activity in endothelial cells. A Notch antagonist, called Notch1 decoy, blocked VEGF activation of Notch/CSL signaling, VEGF-induced HUVEC morphogenesis on both collagen and fibrin gels, and VEGF-induced fibrinolysis. Notch signaling upregulated the expression of MMP9 and MT1-MMP, and activated MMP2 and MMP9 in endothelial cells. Accordingly, we found that the Notch1 decoy-mediated suppression of HUVEC morphogenesis occurred via inhibition of MMP activity. Finally, Notch1 decoy suppressed endothelial MMP9 expression in an
Methods
Reagents and Expression Vectors (adenovirus/retrovirus)
GM6001 was used at 50 μM (Elastin Products Company). eACA was used at 10 mM (Sigma). SU5416 is from Eisai Co. LTD. Notch1 decoy (N1decoy) encodes the extracellular domain of rat Notch1 (bp 241-4229, accession# X57405) fused in frame to human IgG Fc, as described [26]. The constitutively active Notch1 adenovirus (N1IC) encodes the cytoplasmic domain of human Notch1 as described [27]. LacZ, human VEGF165, and Notch1 decoy cDNAs were engineered into pAdlox, recombinant adenoviruses generated and stocks produced as described [28].
Cells and Adenoviral Infections
HUVEC were isolated from human umbilical vein as described [29] and grown using EGM-2 Bullet kit (complete medium, LONZA). Porcine type I collagen is from Nitta Gelatin (Osaka, Japan). Fibrin gels were made by combining 2 mg/ml fibrinogen (Sigma) and 0.0625 U/ml thrombin (Sigma) at 4°C, followed by a one-hour incubation at 37°C. Adenovirus infections were as described [30] at indicated dosage (expressed as MOI - multiplicity of infection).
Western Blotting
HUVEC were cultured on type I collagen-coated plates for 5 days in complete media, then starved in serum free media (SFM, Invitrogen) for 48 hours and cell lysates collected in TENT buffer (50 mM Tris pH8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100). Westerns were conducted using an anti-MT1-MMP antibody (Ab-1, EMD Biosciences).
RT-PCR
HUVEC were seeded on type I collagen gels 2 days after infection and cultured post-confluence for 5 days before isolation of total RNA. Total RNA was isolated with RNeasy mini kit (Qiagen) and First-strand cDNA was synthesized using SuperScript II™ (Invitrogen). PCR primers were designed to recognize transcripts for GAPDH, Jagged1, Dll4, Notch1, Notch4, MMP2, MMP9, MT1-MMP and MT2-MMP. PCR used Platinum Taq DNA polymerase (Invitrogen) and reactions removed at noted cycle number and product analyzed as described [30]. The following oligos were used for RT-PCR:
MT1-MMP
(5'-CGCTACGCCATCCAGGGTCTCAAA-3', 5'-CGGTCATCATCGGGCAGCACAAAA-3'),
MT2-MMP
(5'-TCGACGAAGAGACCAAGGAG-3',
5'-ACTGCCACCAGGAAGAGGTT-3'),
MMP2
(5'-GGGACAAGAACCAGATCACATAC-3',
5'-CTTCTCAAAGTTGTAGGTGGTGG-3'),
MMP9
(5'-GTATTTGTTCAAGGATGGGAAGTAC-3', 5'-GCAGGATGTCATAGGTCACGTAG-3'),
GAPDH
(5'-CGGAGTCAACGGATTTGGTCGTAT-3', 5'-AGCCTTCTCCATGGTGGTGAAGAC-3').
Luciferase Reporter Assay
HUVEC (3.0×105) were infected with adenovirus encoding human VEGF165(Ad-VEGF) and two days later transfected with Effectene (Qiagen) using 475 ng CSL luciferase reporter (pGA981-6) [31] 25 ng Renilla luciferase pRL-SV40 (Promega, Madison, WI) for normalizing transfection efficiency. The following day Luciferase and Renilla activity was determined with Dual-Luciferase® Reporter Assay System (Promega) and Berthold dual-injection luminometer. Notch1 decoy effects on VEGF-activity were evaluated with reporter assays, as above, after co-transduction of HUVEC with adenovirus encoding Notch1 decoy (Ad-N1ECDFc) and Ad-VEGF. All assays were performed in triplicate.
HUVEC Morphogenesis Assay
HUVEC morphogenesis was assessed via microscopy and scored as cells with single or multiple processes after 5 days of culturing on porcine type I collagen gels, as described [32]. Adenovirus infections were performed 2 days prior to seeding on collagen gels. GM6001 was added to medium 1 hr after HUVEC seeding. Cell number was measured using the Cell Counting Kit-8 (Dojindo Molecular Technologies).
Fibrinolytic Assay
HUVEC were seeded as a monolayer on bovine fibrin gels in complete medium in 24 well plate. After 5 days, wells were incubated with 330 ng/ml Thiazolyl Blue Tetrazolium Bromide (4) (Sigma) for 2 hours. Reagent was removed and wells documented by digital photography. Experiments were performed in triplicate.
Gelatin Zymography
Gelatin Zymography was performed on HUVEC conditioned media as described[33]. HUVEC were infected with Ad-VEGF at a MOI of 40 or co-infected with Ad-VEGF and Ad-N1ECDFc each at a MOI of 40. Adenovirus encoding LacZ (Ad-LacZ) was used to normalize infections to 100 MOI. Adenovirus-transduced-HUVEC were cultured on collagen gels, conditioned medium collected at indicated days for MMP9 and day 4 for MMP2.
Dorsal Air Sac (DAS) Assay
The DAS assay was performed as described [34]. KP1/VEGF121 cells were transduced with Ad-GFP or Ad-Notch1 decoy at 60 MOI and packed into Millipore chambers that were transplanted into a DAS of C57BL/6 mice. Mice were sacrificed four days after implantation, and dorsal skin removed and embedded in OCT. Each group consisted of 3-5 mice, and experiments were done in duplicate. Dermal cross-sections were immunostained as described[26] with antibodies against PECAM (Pharminigen) and MMP9 (ABcam) and visualized and imaged by fluorescent microscopy at 20× magnification.
Results
VEGF-induced HUVEC morphogenesis is mediated by Notch
Notch functions downstream of VEGF in arterial/venous specification [35]. We first determined if VEGF induced Notch signaling
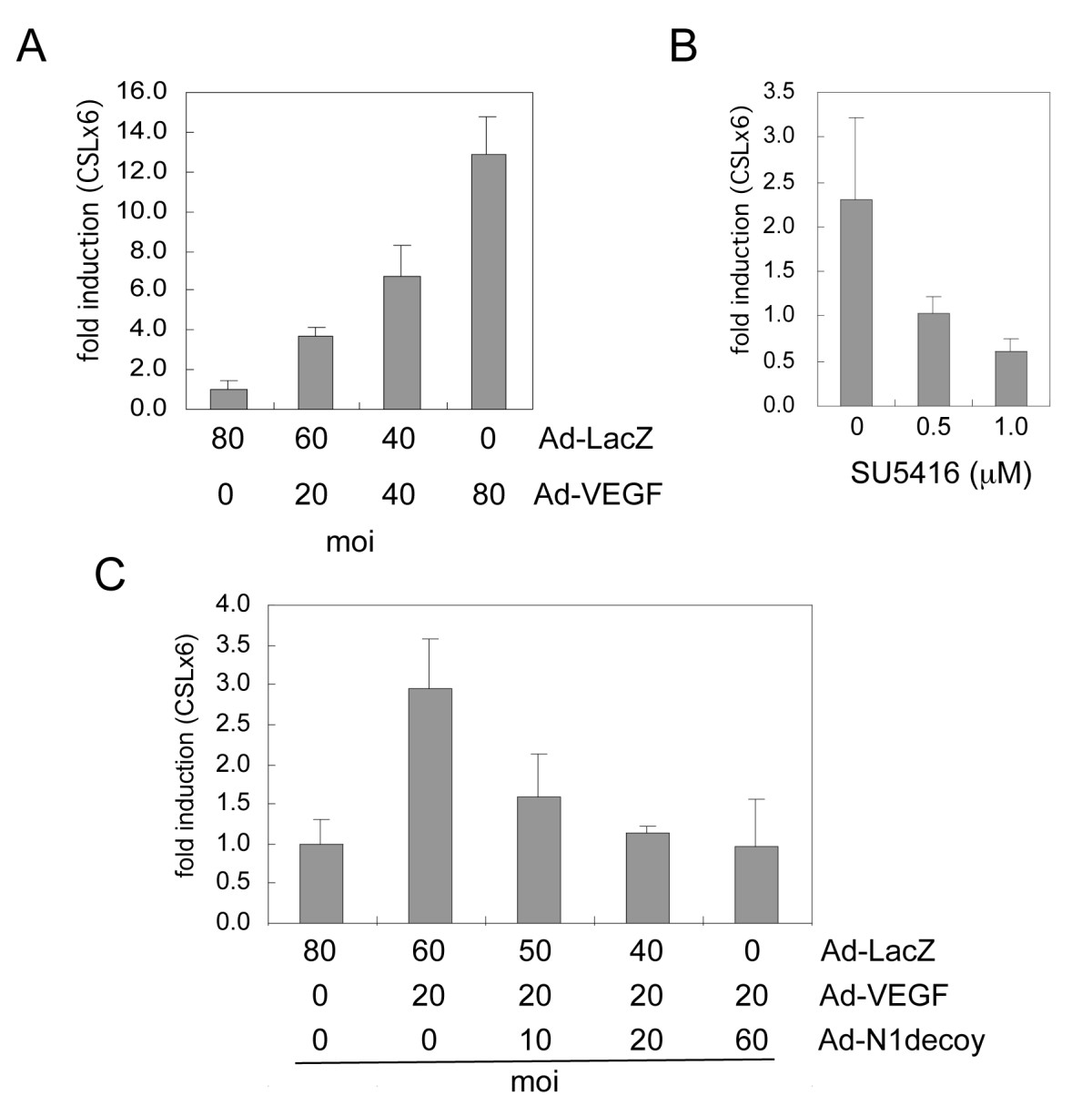
Figure 1
Figure 1 caption
VEGF activated Notch signaling in HUVEC. A) HUVEC were transduced with increasing amounts of Ad-VEGF as indicated, and Notch/CSL reporter activity determined. B) The VEGFR inhibitor (SU5416) perturbed Notch signal activation downstream of VEGF. HUVEC were transduced with Ad-VEGF at 40 MOI and increasing amounts of SU5416 added to HUVEC prior to determining CSL reporter activity. C) Notch1 decoy blocked VEGF-induced Notch signal activation. HUVEC were transduced with Ad-VEGF at 20 MOI and increasing amounts of Ad-Notch1 (N1) decoy as indicated. Ad-LacZ was added to transductions to normalize the MOI between conditions. Notch/CSL reporter luciferase data are represented as fold induction relative to Ad-LacZ controls. Experiments were performed in triplicate, and data of a representative experiment presented.
When grown on three-dimensional fibrin or collagen gels, VEGF promotes the formation of cellular extensions that invade the underlying matrix, a phenomenon termed "HUVEC morphogenesis" [39, 40]. To determine if Notch acts downstream of VEGF-induced morphogenesis, HUVEC were transduced with Ad-VEGF or Ad-LacZ with or without Ad-Notch1 decoy. Three days after seeding on type I collagen gels, HUVEC were scored for the presence of cellular extensions and cell number determined. VEGF promoted HUVEC morphogenesis on collagen gels (Figure 2A), consistent with previous reports [26, 32, 39, 40]. The number of HUVEC with cellular extensions was dependent on the dose of VEGF provided by adenoviral transduction (Figure 2B). In this assay, adenoviral co-transduction of Notch1 decoy with VEGF inhibited VEGF-induced HUVEC morphogenesis, with a nominal effect on proliferation (Figure 2A, C). Thus, Notch mediates VEGF-induced HUVEC morphogenesis on type I collagen and fibrin gels (Figure 2 and data not shown).
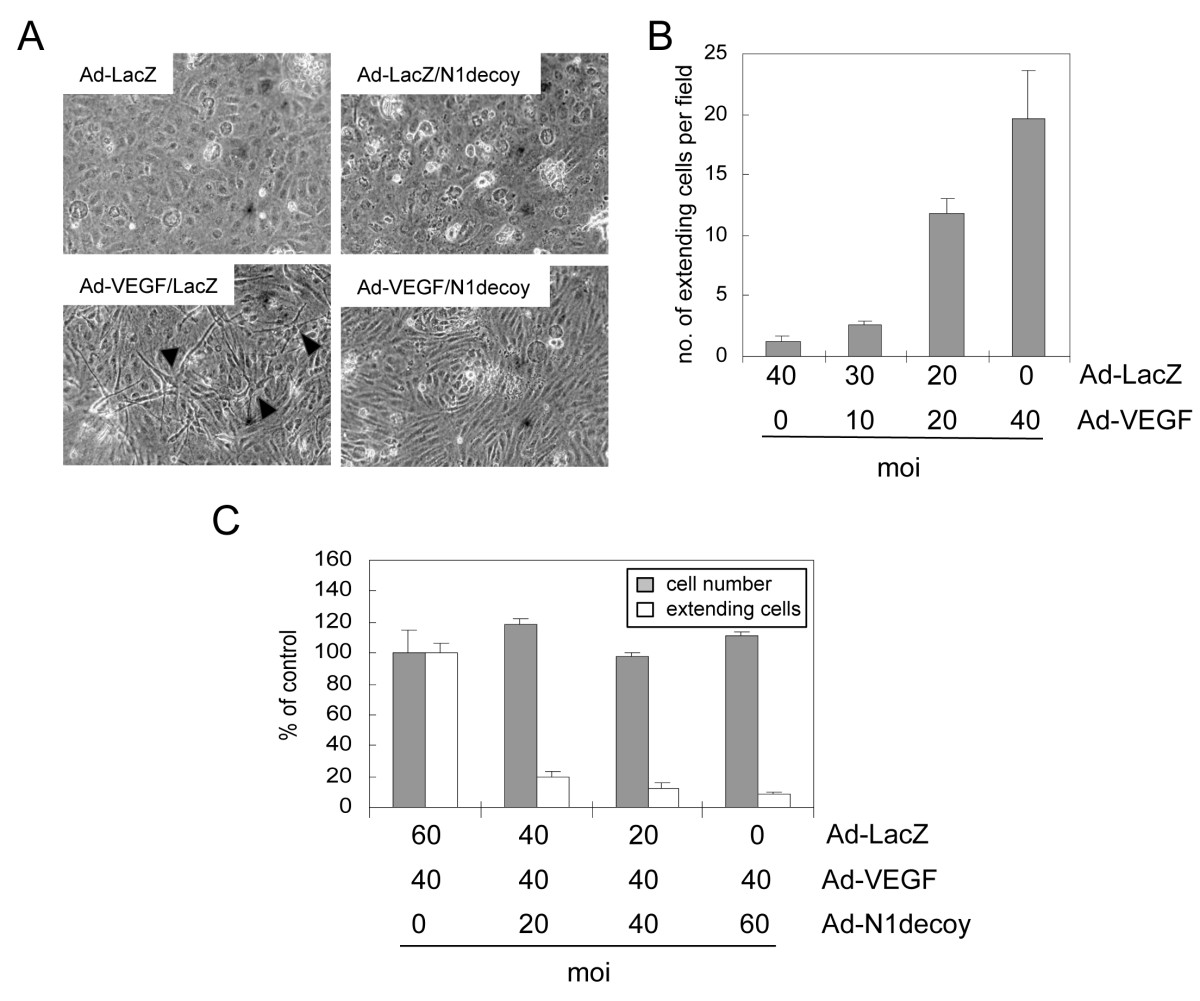
Figure 2
Figure 2 caption
VEGF-dependent HUVEC morphogenic changes are mediated by Notch. A) HUVEC were transduced with Ad-LacZ or Ad-VEGF, with or without Ad-Notch1 (N1) decoy at 40 MOI. Ad-LacZ was used to normalize MOI to 80. VEGF induced HUVEC morphogenesis (lower left panel) that was blocked by N1 decoy (lower right panel). B) Quantification of Ad-VEGF-induced morphogenesis, measured as numbers of HUVEC with cellular extensions. HUVEC were transduced with increasing amounts of Ad-VEGF as indicated and Ad-LacZ was used to normalize MOI to 40. C) N1 decoy blocked VEGF-induced HUVEC morphological changes without affecting total cell number. HUVEC were transduced with Ad-VEGF at 40 MOI and increasing amounts of Ad-N1 decoy as indicated. Ad-LacZ was used to normalize the MOI to 100. Experiments were performed in triplicate, and data of a representative experiment presented.
Notch mediates VEGF-induced fibrinolysis
We next determined if VEGF-mediated ECM degradation occurred via a Notch dependent mechanism. HUVEC were infected with Ad-LacZ or Ad-VEGF with or without Ad-Notch1 decoy and seeded on top of fibrin gels. Six days later, the fibrin gels were incubated with Thiazolyl Blue Tetrazolium Bromide and visualized. Expression of VEGF resulted in the complete degradation of the fibrin gel (Figure 3A). Co-expression of Notch1 decoy with VEGF blocked the fibrinolysis, suggesting that Notch mediates VEGF-induced fibrinolysis in HUVEC.
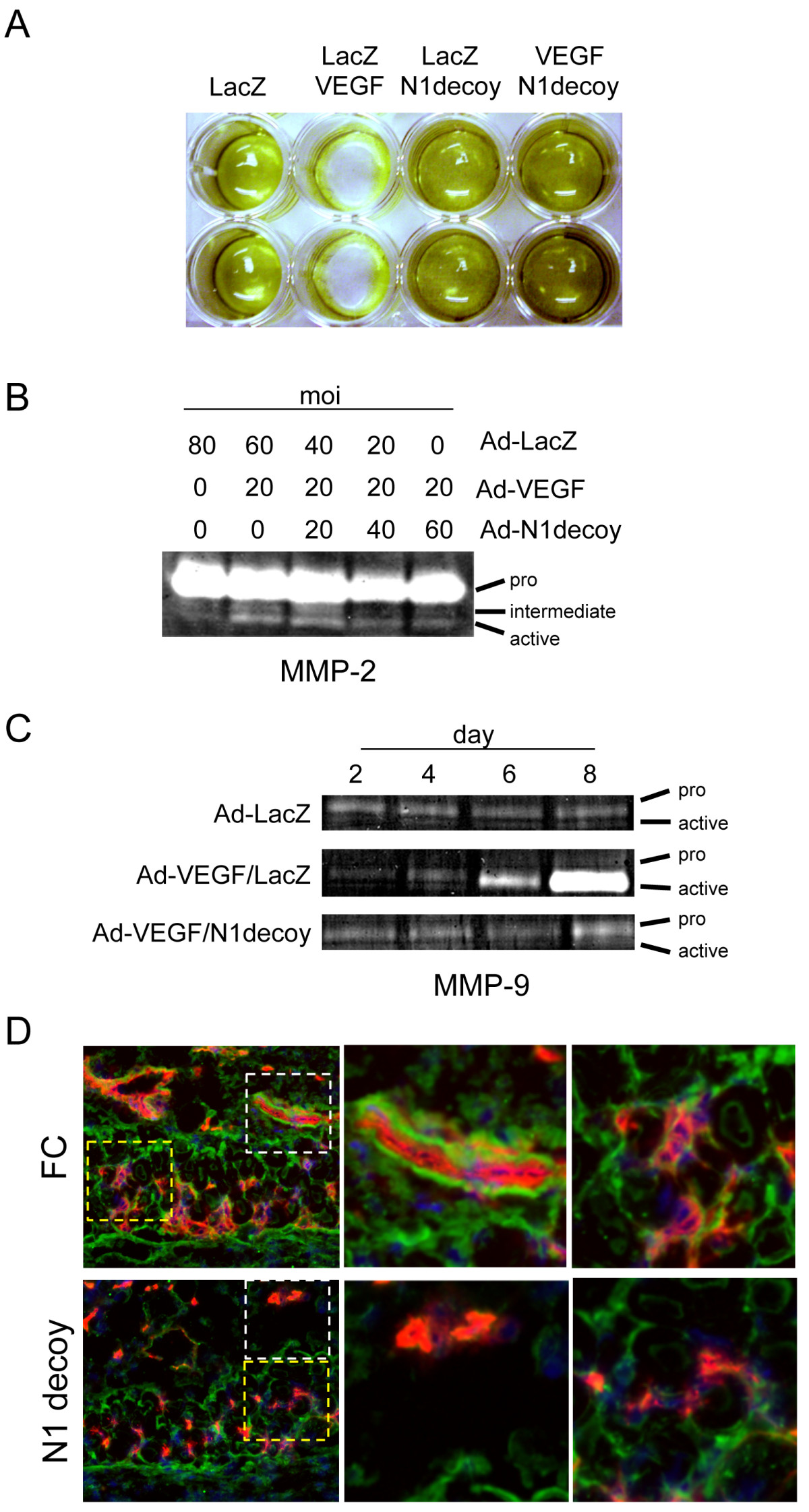
Figure 3
Figure 3 caption
Notch mediates VEGF-induced fibrinolysis and gelatinase activity. A) N1 decoy blocked VEGF-induced fibrinolytic activity. HUVEC were transduced with Ad-VEGF, Ad-N1decoy or both at 40 MOI and Ad-LacZ was used as a control and to normalize the MOI to 80. Transductants were seeded on fibrin gels and fibrin degradation determined after 5 days. B) N1decoy blocked VEGF-induced MMP2 activation. HUVEC were transduced with Ad-VEGF at 20 MOI and increasing amounts of Ad-N1decoy as indicated. Ad-LacZ was used as a control and to normalize the MOI to 80. Four days post-infection, supernatants were collected and subjected to gelatinase zymography. C) N1 decoy blocked VEGF-induced expression and activation of MMP9. HUVEC were transduced with Ad-VEGF at 40 MOI, with or without Ad-N1decoy at 40 MOI and Ad-LacZ was used as a control and to normalize the MOI to 80. Supernatants from transduced HUVEC were collected at 2, 4, 6, and 8 days post-infection and subjected to gelatinase zymography. D) Cross-section of skin from DAS assay stained for PECAM (red) and MMP9 (green). White boxes highlight the large vessels within the subcutaneous adipose depot. Yellow boxes highlight the capillaries within the dermal smooth muscle cell layer. Representative photographs are shown.
Notch regulates VEGF-induced matrix metalloprotease activity
The gelatinases MMP2 and MMP9 function to degrade both fibrin and collagen matrices produced by endothelial cells [41]. VEGF signaling promotes the secretion and activation of MMPs, such as MMP2 and MMP9 [42]. As Notch signaling mediated VEGF-induced HUVEC morphogenesis, we determined if the induction of MMP2 and MMP9 activity by VEGF was mediated by Notch. HUVEC were infected with Ad-VEGF and increasing amounts of Ad-Notch1 decoy, and conditioned media was used in zymographic analysis. After four days, the level of MMP2 pro-form was unchanged, while VEGF converted MMP2 to an active form (Figure 3B). VEGF-induced MMP9 activity 4 days post-transduction, and this activity increased over the following four days (Figure 3C). The VEGF-mediated MMP activation we observed was consistent with previous reports [42]. To examine the role of Notch in the VEGF induced MMP activities, we assessed MMPs after expression of the Notch1 decoy. Co-expression of VEGF with Notch1 decoy inhibited the activation of MMP2 and blocked the transcriptional induction and biochemical activation of MMP9 by VEGF (Figure 3B, C). Thus, VEGF activated MMP2 and MMP9 gelatinase activity by a Notch-dependent mechanism.
To determine if Notch influenced MMP9 expression during VEGF-induced angiogenesis, we evaluated the expression of MMP9
Notch signaling induces MMP9 and MT1-MMP expression in HUVEC
Since inhibition of Notch signaling abrogated VEGF induction of MMP2 and MMP9 activity, we next examined whether Notch activation upregulated the expression of MMP2 and MMP9 in HUVEC. HUVEC were infected with an adenovirus encoding a constitutively active cytoplasmic form of Notch1 (N1IC), RNA isolated and RT-PCR performed for MMP2, MMP9, and GAPDH. Notch1 activation upregulated MMP9 transcripts (Figure 4A). Though we found that Notch1 decoy blocked VEGF from activating MMP2 (Figure 3B), increased Notch1 signaling paradoxically suppressed MMP2 transcript levels (Figure 4A). On the cell surface, MMP2 is activated by membrane-type MMPs (MT-MMPs) [7]; therefore, we determined if Notch signaling altered the expression of MT-MMPs, MT1-MMP and MT2-MMP. Expression of the activated form of Notch1 upregulated both MT1-MMP transcripts and protein, but had no affect on MT2-MMP (Figure 4B).
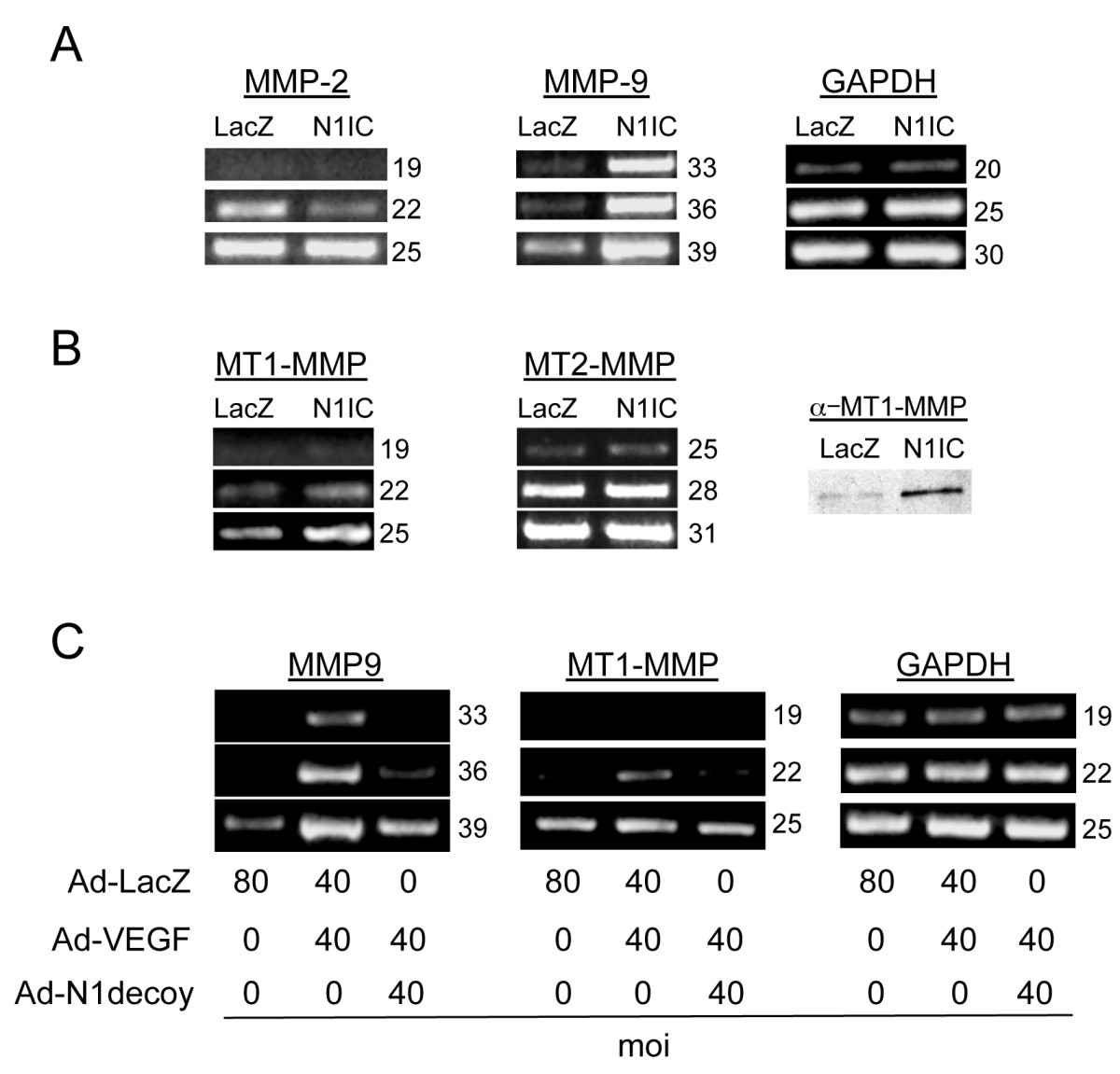
Figure 4
Figure 4 caption
Notch induced MMP9 and MT1-MMP expression downstream of VEGF. A) Transduction of an activated form of Notch1 (N1IC) induced MMP9 transcripts in HUVEC. RT-PCR analysis of MMP2 and MMP9 compared to control GAPDH. B) Transduction of N1IC induced MT1-MMP. RT-PCR analysis of MT1-MMP and MT2-MMP (left panels) compared to control GAPDH (see A). Western analysis of MT1-MMP (right panel). C) N1decoy blocked VEGF-mediated induction of MMP9 and MT1-MMP determined by RT-PCR; GAPDH serves as a control. HUVEC were transduced with Ad-VEGF at 40 MOI, with or without Ad-N1decoy at 40 MOI and Ad-LacZ was used as a control and to normalize the MOI to 80. For RT-PCR experiments, the number of PCR cycles is indicated to the right of the figures. Experiments were performed in triplicate, and data of a representative experiment presented.
Next, we asked whether Notch activity is required for the VEGF-induction of MMP9 and MT1-MMP. Ectopic expression of VEGF increased both MMP9 and MT1-MMP transcripts (Figure 4C). Co-infections of Ad-Notch1 decoy with Ad-VEGF suppressed the expression of both MMP9 and MT1-MMP transcripts. Taken together, these data demonstrate that Notch1 activation increased MMP2 and MMP9 activity. Thus, Notch functions downstream of VEGF to regulate MMPs via distinct mechanisms; Notch upregulated MMP9 activity at the transcriptional level, while it regulated MMP2 activity at the cell surface via the induction of MT1-MMP.
Notch-induced HUVEC morphogenesis is mediated by MMPs
Since VEGF-induced HUVEC morphogenesis requires Notch signal activation, we determined whether Notch mediates this process via MMP activation. HUVEC were transduced with Ad-LacZ or Ad-N1IC and seeded on type I collagen or fibrin gels. After 7 days, the presence of HUVEC with cell extensions was evaluated and quantified. We used an adenovirus that co-expressed N1IC and GFP to visualize the cells expressing the activated form of Notch1. Notch1 activation induced HUVEC morphological changes (Additional File 2), similar to that observed for ectopic VEGF (Figure 2A). In this assay, Notch1 functioned in a cell autonomous fashion, as Notch-activated cells (identified as GFP positive cells) displayed cellular extensions when compared to GFP negative cells. Moreover, the number of HUVEC undergoing morphological changes increased with higher doses of Ad-N1IC adenovirus (data not shown). Using this assay, we asked if Notch promoted HUVEC morphogenesis via the induction and activation of MMPs. Ad-LacZ and Ad-N1IC-transduced HUVEC were seeded on either type I collagen or fibrin gels and the MMP inhibitor GM6001 introduced to the media. GM6001 is an inhibitor that targets multiple MMPs, including MMP2, MMP9, and MT1-MMP [44]. Notch-mediated HUVEC morphogenesis was inhibited by GM6001 when assayed on either collagen (Figure 5A, B) or fibrin gels (Figure 5C,D). As fibrinolysis also involves the uPA/PAR pathway, we introduced the serine-protease inhibitor, eACE, to block this pathway in Ad-infected HUVEC grown on fibrin gels (Figure 5C, D). The serine-protease inhibitor did not block Notch induced HUVEC morphogenesis, and did not have an additive affect when used in combination with the MMP inhibitor GM6001 (Figure 5D). Thus, VEGF requires Notch signaling to regulate both expression and activation of specific MMPs, and Notch, in turn, utilizes MMP activation to promote HUVEC morphogenesis.
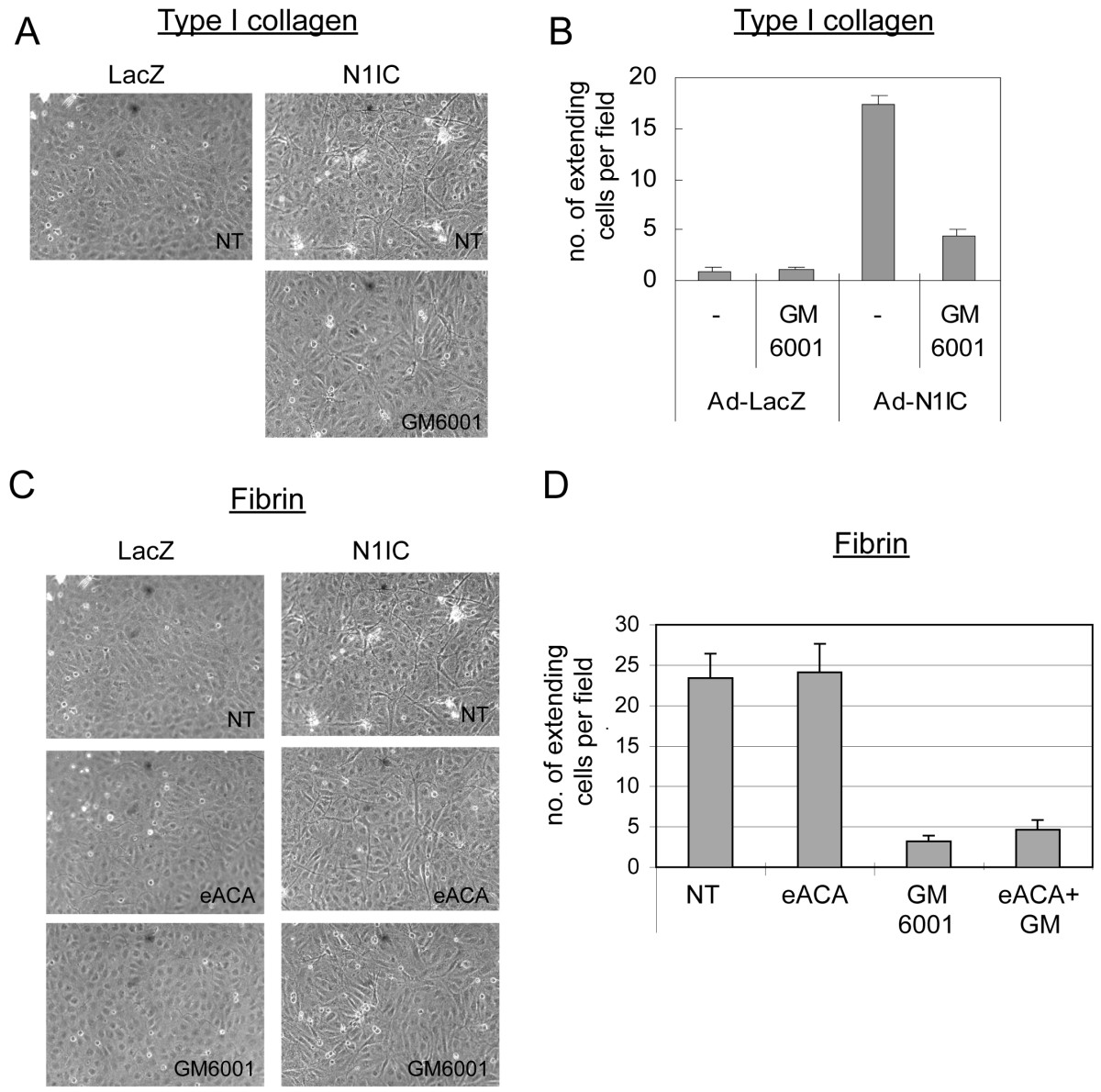
Figure 5
Figure 5 caption
Notch-induced HUVEC morphogenesis was mediated via its induction of MMPs. A) Notch-induced HUVEC morphogenesis on collagen was blocked by addition of an MMP inhibitor. HUVEC were transduced with either Ad-LacZ or Ad-N1IC at 40 MOI and seeded on type 1 collagen gels in the absence or presence of 50 μM GM6001. Images are representative of 5 days post-infection. B) Quantification of Ad-N1IC-induced morphogenesis on collagen, measured as numbers of HUVEC with cellular extensions. C) Notch-induced HUVEC morphogenesis on fibrin was blocked by addition of an MMP inhibitor, but not a serine-protease inhibitor. HUVEC were transduced with either Ad-LacZ or Ad-N1IC at 40 MOI and seeded on fibrin gels in the absence or presence of the matrix metalloprotease inhibitor (50 μM GM6001), or the serine-protease inhibitor, (10 mM eACE). Images are representative of 5 days post-infection. D) Quantification of Ad-N1IC-induced morphogenesis on fibrin. Experiments were performed in triplicate, and data of a representative experiment presented. NT (No Treatment).
Discussion
In this study, we describe a new means by which Notch functions downstream of VEGF to regulate the angiogenic process. We identified MMPs as novel targets of Notch signaling in endothelial cells. Our data suggest that VEGF activates Notch signaling by upregulating the expression of Dll4 and Notch4, consistent with other reports [22]. Notch1 decoy blocked VEGF-activation of Notch/CSL signaling, VEGF-induced HUVEC morphogenesis on collagen and fibrin gels, VEGF-dependent fibrinolysis and VEGF-induced dermal angiogenesis
Pericellular matrix metalloproteases regulate the local availability of bioactive angiogenic factors, such as VEGF and bFGF. It has been proposed that matrix metalloproteases function to increase VEGF activity to promote pathological angiogenesis during tumorigenesis [42]. Consistent with this hypothesis, deletion of MMP9 in a mouse model of pancreatic β-cell carcinoma, Rip1Tag2, suppressed tumor progression [47]. In these mice, the level of VEGF was unaltered, but mobilization of VEGF from the extracellular matrix was inhibited. Thus, endothelial upregulation of MMP9 by Notch may lead to the localized release and activation of matrix-bound growth factors.
Besides its role in processing and activating MMP2, MT1-MMP also functions to degrade the surrounding fibrin matrix during physiological and pathological angiogenesis. The breakdown of the local fibrin matrix is essential in the initiation and propagation of angiogenic responses, and generates a provisional matrix that can sustain the formation of nascent vessels during wound healing, inflammation and tumor growth. MT1-MMP localizes at the leading edge of migrating endothelial cells and is thought to aid in the degradation of the extracellular matrix to facilitate endothelial cell invasion [48]. Although MT1-MMP may be the most potent fibrinolytic MMP, the gelatinases MMP2 and MMP9 also recognize fibrin as a substrate. More recently, Stratman et al demonstrated that the formation of vascular guidance tunnels within collagen gels occurred via a MT1-MMP-dependent proteolytic process and that these tunnels become conduits for EC motility in sprouting angiogenesis [13] Thus, the induction of MT1-MMP by Notch may lead to the activation of MMP2 that together with MT1-MMP and MMP9 promote extracellular matrix degradation and endothelial cell morphogenesis.
MT1-MMP activity has also been shown to be critical for endothelial migration during sprouting angiogenesis [42, 49]; however Notch signal activation is a potent inhibitor of endothelial cell migration [50, 51], which suggests that Notch signaling has both anti- and pro-angiogenic functions. Thus, Notch may have an early pro-angiogenic function in the induction and activation MMPs to promote the degradation of the pericellular matrix, but it subsequently needs to be turned off to allow for endothelial cell migration and tube formation to progress. MT1-MMP activity has also been shown to be critical for endothelial lumen formation [13]. Taking this finding into consideration, Notch may be turned off in tip cells and induces MMPs in stalk cells, which may be important for remodeling the matrix during the process of lumen formation. The latter model fits well with our current understanding of Notch function in the restriction of endothelial sprouting by blocking tip cell differentiation [52].
The molecular mechanism by which Notch regulates MMP9 and MT1-MMP expression in HUVEC is unclear. In lung metastases of Rip1Tag2 mice, MMP9 expression, within the endothelium, was regulated by VEGFR-1 [53]. Since Notch induces VEGFR-1 expression in endothelial cells [43, 54], VEGFR-1 signaling may mediate the induction of MMP9 by Notch. However, Notch1 has also been shown to regulate the expression and activation of MMP9 through NF-κB in pancreatic cancer cells [55], suggesting that the regulation of MMP9 expression and activity in endothelial cells by Notch may be mediated by NF-κB signaling. Finally, the activation of MMP9 at the cell surface is dependent on the uPA/plasmin pathway. In human prostatic cancer cells, knock-down of Notch1 resulted in a downregulation of MMP9 and uPA and its receptor, uPAR [56]. From our data it is unclear whether Notch regulates MMP9 solely at the level of transcription, and the possibility remains that it may regulate the uPA/plasmin pathway to activate MMP9 at the cell surface. This is a subject of future investigation.
We propose that Notch activates MMP2 via induction of MT1-MMP, which is present in its active form at the cell surface. In the tumor microenvironment, an imbalance may arise from an increase in MMP expression/activity and a decrease in the expression of the MT-MMP inhibitors, TIMP-2 or TIMP-3 [4, 5]. This imbalance would result in a pro-angiogenic state. A similar imbalance in MMP/TIMP activity ratio has also been implicated in aortic aneurysm, varicose veins, hypertension and preeclampsia [6]. As inhibition of Notch signaling with Notch1 decoy blocked the induction of MMP9 and MT1-MMP transcripts in HUVEC, blocking Notch signaling in pathological settings may perturb angiogenesis and may prove therapeutically useful in the treatment of vascular disorders.
Additional files
Additional file 1: VEGF induced Notch and Notch ligand expression in HUVEC. HUVEC were transduced with either Ad-LacZ or Ad-VEGF at 40 MOI. Two days later, total RNA was isolated and RT-PCR performed with PCR primers designed to amplify GAPDH, Notch1, Notch4, Jagged1, Dll4. Reactions were removed at noted cycle number and product analyzed as described [30]. Number of PCR cycles is indicated. (TIFF 3 MB)
Additional file 2: Notch signal activation cell autonomously altered HUVEC morphology. HUVEC were transduced with an adenovirus which co-expresses N1IC and GFP at 40 MOI. N1IC/GFP expressing tranductants were mixed with control HUVEC and cultured on type 1 collagen gels for 7 days. Notch activated cells, identified as GFP positive cells, display cellular extension, as compared to GFP negative cells. (TIFF 3 MB)
Acknowledgements
Supported by NIH 5RO1HL062454 (JK), NIH 5R01CA136673 (JK, CJS.) and from the Department of Defense W81XWH-07-1-0362 (J.K.). We thank Valeriya Borisenko for technical assistance.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2
Authors’ original file for figure 3
Authors’ original file for figure 4
Authors’ original file for figure 5
References
- Influence of VEGF-R2 inhibition on MMP secretion and motility of microvascular human cerebral endothelial cells (HCEC). J Neurooncol. 2003;62:221-231.
- Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511-521.
- Impaired angiogenesis in SHR is associated with decreased KDR and MT1-MMP expression. Biochem Biophys Res Commun. 2004;315:363-368.
- Pleiotropic roles of matrix metalloproteases in tumor angiogenesis: contrasting, overlapping and compensatory functions. Biochim Biophys Acta. 2010;1803:103-120.
- Matrix metalloproteases: regulators of the tumor microenvironment. Cell. 2010;141:52-67.
- Acute actions and novel targets of matrix metalloproteases in the heart and vasculature. Br J Pharmacol. 2007;152:189-205.
- Seiki M, Mori H, Kajita M, Uekita T, Itoh Y: Membrane-type 1 matrix metalloprotease and cell migration. Biochem Soc Symp. 2003: 253-262.
- Membrane-type matrix metalloproteases (MT-MMPs) in tumor metastasis. J Biochem. 1996;119:209-215.
- Membrane-type matrix metalloprotease 1 is a gelatinolytic enzyme and is secreted in a complex with tissue inhibitor of metalloproteases 2. Cancer Res. 1996;56:2707-2710.
- MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004;167:757-767.
- Navigating ECM barriers at the invasive front: the cancer cell-stroma interface. Annu Rev Cell Dev Biol. 2009;25:567-595.
- MT1-MMP- and Cdc42-dependent signaling co-regulate cell invasion and tunnel formation in 3D collagen matrices. J Cell Sci. 2009;122:4558-4569.
- Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood. 2009;114:237-247.
- Notch function in the vasculature: insights from zebrafish, mouse and man. Bioessays. 2004;26:225-234.
- Notch: a membrane-bound transcription factor. J Cell Sci. 2002;115:1095-1097.
- The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216-233.
- Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci USA. 2001;98:5643-5648.
- Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439-442.
- Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci USA. 2004;101:15949-15954.
- Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004;18:2469-2473.
- Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18:2474-2478.
- Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol Cell Biol. 2003;23:14-25.
- Up-regulation of the Notch ligand Delta-like 4 inhibits VEGF-induced endothelial cell function. Blood. 2006;107:931-939.
- Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444:1032-1037.
- Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083-1087.
- A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res. 2008;68:4727-4735.
- Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001;20:3427-3436.
- Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997;71:1842-1849.
- Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest. 1973;52:2745-2756.
- Notch signaling in primary endothelial cells. Ann N Y Acad Sci. 2003;995:162-170.
- Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development. 1997;124:4133-4141.
- Notch oncoproteins depend on gamma-secretase/presenilin activity for processing and function. J Biol Chem. 2004;279:30771-30780.
- Characterization of rat aortic fragment within collagen gel as an angiogenesis model; capillary morphology may reflect the action mechanisms of angiogenesis inhibitors. Biol Pharm Bull. 2002;25:446-451.
- Establishment of a quantitative mouse dorsal air sac model and its application to evaluate a new angiogenesis inhibitor. Oncol Res. 1999;11:319-329.
- sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell. 2002;3:127-136.
- Functional replacement of the intracellular region of the Notch1 receptor by Epstein-Barr virus nuclear antigen 2. J Virol. 1998;72:6034-6039.
- Antiangiogenic effect by SU5416 is partly attributable to inhibition of Flt-1 receptor signaling. Mol Cancer Ther. 2002;1:295-302.
- A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. J Clin Invest. 2007;117:2477-2485.
- Phorbol esters induce angiogenesis in vitro from large-vessel endothelial cells. J Cell Physiol. 1987;130:284-291.
- Cooperative effect of TNFalpha, bFGF, and VEGF on the formation of tubular structures of human microvascular endothelial cells in a fibrin matrix. Role of urokinase activity. J Cell Biol. 1996;132:1177-1188.
- Extracellular proteolysis and angiogenesis. Thromb Haemost. 2001;86:346-355.
- Role of the matrix metalloprotease and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21:1104-1117.
- Notch regulates the angiogenic response via induction of VEGFR-1. J Angiogenes Res. 2010;2:3-.
- Inhibition of membrane-type 1 matrix metalloprotease by hydroxamate inhibitors: an examination of the subsite pocket. J Med Chem. 1998;41:1209-1217.
- Matrix metalloproteases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9-34.
- Tumstatin, the NC1 domain of alpha3 chain of type IV collagen, is an endogenous inhibitor of pathological angiogenesis and suppresses tumor growth. Biochem Biophys Res Commun. 2005;333:292-298.
- Matrix metalloprotease-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737-744.
- Roles of pericellular proteolysis by membrane type-1 matrix metalloprotease in cancer invasion and angiogenesis. Cancer Sci. 2003;94:569-574.
- Dissecting the role of matrix metalloproteases (MMP) and integrin alpha(v)beta3 in angiogenesis in vitro: absence of hemopexin C domain bioactivity, but membrane-Type 1-MMP and alpha(v)beta3 are critical. Cancer Res. 2005;65:9377-9387.
- Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and downregulates VEGFR-2/KDR expression. Microvasc Res. 2002;64:372-383.
- The role of the vascular endothelial growth factor-Delta-like 4 ligand/Notch4-ephrin B2 cascade in tumor vessel remodeling and endothelial cell functions. Cancer Res. 2006;66:8501-8510.
- Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196-208.
- MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell. 2002;2:289-300.
- Regulation of multiple angiogenic pathways by Dll4 and Notch in human umbilical vein endothelial cells. Microvasc Res. 2008;75:144-154.
- Down-regulation of notch-1 inhibits invasion by inactivation of nuclear factor-kappaB, vascular endothelial growth factor, and matrix metalloprotease-9 in pancreatic cancer cells. Cancer Res. 2006;66:2778-2784.
- Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-kappaB signaling pathways. J Cell Biochem. 2010;109:726-736.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.