Cyclic strain upregulates VEGF and attenuates proliferation of vascular smooth muscle cells
Vascular Cell. 2011;
Received: 27 May 2011 | Accepted: 19 September 2011 | Published: 19 September 2011
Vascular Cell ISSN: 2045-824X
Abstract
Objective
Vascular smooth muscle cell (VSMC) hypertrophy and proliferation occur in response to strain-induced local and systemic inflammatory cytokines and growth factors which may contribute to hypertension, atherosclerosis, and restenosis. We hypothesize VSMC strain, modeling normotensive arterial pressure waveforms in vitro, results in attenuated proliferative and increased hypertrophic responses 48 hrs post-strain.
Methods
Using Flexcell Bioflex Systems we determined the morphological, hyperplastic and hypertrophic responses of non-strained and biomechanically strained cultured rat A7R5 VSMC. We measured secretion of nitric oxide, key cytokine/growth factors and intracellular mediators involved in VSMC proliferation via fluorescence spectroscopy and protein microarrays. We also investigated the potential roles of VEGF on VSMC strain-induced proliferation.
Results
Protein microarrays revealed significant increases in VEGF secretion in response to 18 hours mechanical strain, a result that ELISA data corroborated. Apoptosis-inducing nitric oxide (NO) levels also increased 43% 48 hrs post-strain. Non-strained cells incubated with exogenous VEGF did not reproduce the antimitogenic effect. However, anti-VEGF reversed the antimitogenic effect of mechanical strain. Antibody microarrays of strained VSMC lysates revealed MEK1, MEK2, phospo-MEK1T385, T291, T298, phospho-Erk1/2T202+Y204/T185+T187, and PKC isoforms expression were universally increased, suggesting a proliferative/inflammatory signaling state. Conversely, VSMC strain decreased expression levels of Cdk1, Cdk2, Cdk4, and Cdk6 by 25-50% suggesting a partially inhibited proliferative signaling cascade.
Conclusions
Subjecting VSMC to cyclic biomechanical strain in vitro promotes cell hypertrophy while attenuating cellular proliferation. We also report an upregulation of MEK and ERK activation suggestive of a proliferative phenotype. Hhowever, the proliferative response appears to be aborogated by enhanced antimitogenic cytokine VEGF, NO secretion and downregulation of Cdk expression. Although exogenous VEGF alone is not sufficient to promote the quiescent VSMC phenotype, we provide evidence suggesting that strain is a necessary component to induce VSMC response to the antimitogenic effects of VEGF. Taken together these data indicate that VEGF plays a critical role in mechanical strain-induced VSMC proliferation and vessel wall remodeling. Whether VEGF and/or NO inhibit signaling distal to Erk 1/2 is currently under investigation.
Keywords
blood vessels cell proliferation biomechanical strain VEGF vascular smooth muscle cytokines nitric oxideIntroduction
Cyclic strain (CS) induced by changes in blood pressure can regulate vascular remodeling, proliferation, apoptosis, cell phenotypic changes, and secretion of extracellular matrix proteins and cytokines [1]. Pathological states such as hypertension, atherosclerosis, and restenosis lead to further changes in vascular remodeling [1]. Vascular smooth muscle cells (VSMC) are prevalent in vessel walls and are mechanotransducers of strain and shear stress [2]. In vitro biophysical strain models that mimic arterial pressure waveforms have provided important mechanistic explanations about VSMC responses [2, 3]. For instance, under normotensive conditions, hemodynamic forces strain large arteries up to 10% (termed "physiological" strain; [4]), which shifts VSMC from a synthetic/secretory phenotype in static culture to a non-proliferative contractile phenotype [5–7]. Further elevation of strain magnitude (15 - 30%) [4] shifts cells to a contractile proliferative phenotype [7].
Mechanotransduction cascades have been implicated in the regulation of the VSMC proliferative response [2] including VSMC-derived cytokines such as IGF-1 and PDGF [8–10]. Vascular endothelial growth factor (VEGF) is also an important repressor of VSMC proliferation [11, 12], and is upregulated in aorta from hypertensive rats [13] and in strained VSMC cultures [14]. VSMC calcium homeostasis also plays a pivotal role in strain-induced VSMC proliferation which is dependent upon both stretch activated calcium channels (SACC) and voltage operated calcium channels [15]. Thus the proliferative effects of biomechanical strain on VSMC are dependent upon the complex interplay of the intracellular signaling cascades involved in proliferation and progression of the cell cycle.
The current study investigates the antimitogenic effects of a 10% CS on VSMC. We assessed strain-regulation of autocrine cytokine and growth factor secretion and hypothesized that VEGF plays an important role in mediating strain-induced antimitogenic behavior. We also examined the potential role of SACCs in mediating this antiproliferative effect and the expression and phosphorylation of the mechanosensitive ERK1/2 signaling cascade, PKC isoforms, and Cdk isoforms.
Materials and methods
Vascular Smooth Muscle Cell Cultures
A7r5 rat thoracic aortas VSMC were purchased from American Type Culture Collection (Rockville, MD). Cells were cultured in Dulbecco's modified Eagles Medium (DMEM) supplemented with 9% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C, 5% CO2, and 100% humidity. Growth medium (GM) was replaced every other day, and upon 80% confluency cells were passed at a ratio of 1 to 4 (acquired in 4 to 5 days). All experiments utilized passage-matched VSMC between passage 2 and 10.
In Vitro Strain Apparatus
The Flexercell FX-4000 Tension Plus (Flexcell International Corp, Hillsborough, NC) is a computer-based system which utilizes vacuum suction to strain cells seeded on flexible collagen I-coated electrometric membranes. The deformation of the elastomeric membrane causes the adherent cells to similarly deform. The strain profile was created by programming the magnitude, duration, and frequency of negative pressure to match the desired model. Strain on the cells utilizing this apparatus has been empirically determined to be heterobiaxial [16].
Strain Profile
Cells were seeded (65,000 cells/well) onto 6-well collagen I-coated Bioflex plates (Flexcell International Corp, Hillsborough, NC). Once the cells were ~50-60% confluent (~24 hours after seeding), the GM was replaced with serum-free medium for 24 hours to induce quiescence, and on the day of the experiment the medium was replaced with fresh GM. VSMC were then cyclically strained for 1, 6, 18 or 48 hours. The strain paradigm modeled an aortic pressure waveform and included a modeled dicrotic notch (Figure 1 top). Control cells were grown in the identical environment, but were not subjected to strain.
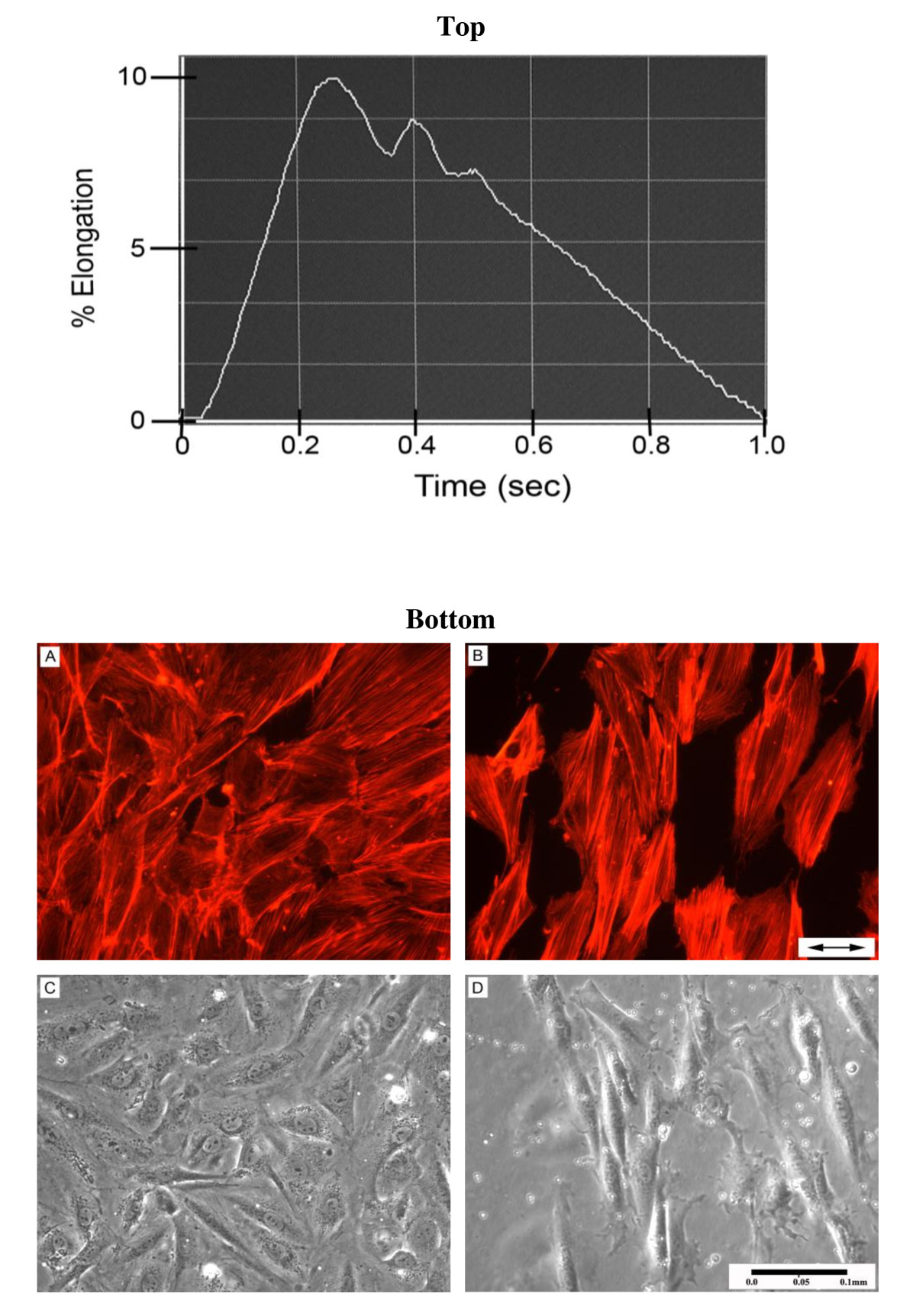
Figure 1
Figure 1 caption
Aortic Pressure Waveform Regulates VSMC Alignment: (Top Panel) Strain profile used in this study. Control cells were not strained. (Bottom Panel) Control VSMC (A, C) and VSMC strained cyclically for 48 hours at 1 Hz, 10% (B, D). Representative wells in each experimental group were fluorometrically stained for assessment of intracellular actin (A, B). Arrows in panel B indicate the direction of the dominant strain vector.
Cell viability, growth assessments, and morphology
A subset of control and strained cells was lysed and cell lysates were used for dsDNA (Invitrogen Corporation; Eugene, OR) and protein quantification (Pierce Chemical Co., Rockford, IL). The ratio of protein to dsDNA was calculated as one proxy of cellular hypertrophy. Phase contrast microscopy at 48 hours was used to quantify cell viability, cell number, cell area and perimeter, and morphologic changes. Actin architecture changes were assessed using rhodamine-conjugated phalloidin as previously described [17]. Photomicrographs were digitally captured using an IX71 Olympus inverted fluorescent microscope and DP71 camera (Olympus America Inc, Melville, NY) and cell area and perimeter measure obtained via Image J v1.37 (National Institute of Health; http://rsb.info.nih.gov/ij/). Cells were randomly selected from four representative experiments and at least four cells from each photographic quadrant were measured in blinded fashion (control: n = 17; strain: n = 16). Cells per high powered field (cells/hpf) were calculated from digital images of control (n = 24) and strained (n = 27) cells from four experiments.
Cytokine Detection
Conditioned media samples obtained from six control and strained samples were collected at the end of 48 hours and frozen at -80°C until assayed. Array membranes were prepared and percent change from control (%CFC) was determined by methods described previously [18]. Further assessment of VEGF was done using a rat enzyme-linked immunosorbent assay (ELISA) kit (Raybiotech Inc., Norcross, GA). VSMC strain-derived NO was quantified using a fluorometric assay kit, (Cayman Chemical Company, Ann Arbor, MI). All data were normalized to cell proliferation data.
Exogenous VEGF Experiments
In a subset of experiments, quiescent medium was replaced with 100 μl of GM containing rat VEGF (R&D Systems, Minneapolis, MN) concentrations of 0, 0.003, 0.03, 0.3, 3, and 30 ng/ml. Cells were then incubated for an additional 18 or 48 hours and proliferation rates at these two time points were measured using the CellTiter 96® Aqueous One Solution Cell Proliferation Assay (MTS). Neither control nor VEGF-treated VSMC were strained in this experimental series. In a separate set of experiments, control and strained VSMC were treated with 0.4 μg/ml of anti-VEGF (R&D Systems, Minneapolis, MN) and assess for cell proliferation by method previously described.
Effects of Extracellular Calcium and Gadolinium
In a subset of experiments, serum-free medium was replaced with one of the following growth media containing 9% FBS: 1) Hyclone custom calcium-free medium (CFM; Thermo Scientific Hyclone AdvanceStem, Logan, UT) with 0.0018 moles/L CaCl2, 2) CFM with 0.0018 moles/L NaCl (osmotic control), and 3.) CFM with 0.0018 moles/L CaCl2 and 30 μM of GdCl3. Cells were then strained or left static for 48 hours.
Intracellular Protein Microarrays
Antibody microarray services from Kinexus Bioinformatics Corporation (Vancouver, BC, Canada) detected the expression and phosphorylation states of 608 cell signaling proteins (in duplicate) after 48 hours strain from 10 culture wells of control and strained VSMC. Samples were prepared per Kinexus protocol; five wells were collectively pooled for each assay based upon the media and protein requirements for detection. Protein spotting and hybridization onto the array, scanning, imaging, and quantitative analysis of the enhanced chemiluminescence signal of the signaling proteins was performed (in a blinded fashion) by Kinexus Bioinformatics Corporation. Only proteins exhibiting a %CFC of ± 25% were considered for further analysis.
Replicates and statistical analysis
Experiments were completed a minimum of three times and had three or more replicate wells per experiment, except for the anti-VEGF and calcium/gadolinium experiments which was completed over two experiments with three or more replicate wells. Kinexus antibody microarray experiments consisted of ten pooled replicate wells and the assay was done in duplicate. All data are expressed as mean ± SEM and means were compared by two-tailed unpaired t-tests or one-sample two-tailed t-tests where appropriate. We tested for polyploidy (dsDNA:cell) by calculating the variance of ratios [19] between the mean ± SEM dsDNA and the mean ± SEM cells per high powered field for both strained and non strained VSMC; p < 0.05 were determined to be significantly different. All data were analyzed with Microsoft Excel (Microsoft Corporation) and Prism 4.03 (GraphPad Software, Inc., San Diego, California).
Results
Intracellular actin and long axes of strain-induced VSMC cytoskeleton aligned nearly perpendicularly to the dominant strain vector (Figure 1; bottom panels B and D). Non-strained VSMC displayed no such alignment characteristics (Figure 1; bottom panels A and C). Strained VSMC also exhibited decreased proliferation and increased cell size versus control. Strained cells had an average of 37% greater area (p < 0.05), 36% larger perimeter (p < 0.001) and 38% fewer cells/hpf (p < 0.001) when compared to control after 48 hours of CS (Figure 2A-C). Double-stranded DNA concentration (Figure 2D) was also reduced by 33% in strained VSMC (p < 0.001). To confirm this dsDNA reduction was not a function of cellular polyploidy as is common among hypertensive VSMC, we calculated the concentration dsDNA:cells/hpf ratio of strained vs. control (0.181 ± 0.016 μg DNA/cell vs. 0.167 ± 0.056 μg DNA/cell) and determined there was no significant difference (p > 0.05). Strained VSMC exhibited an 18% increase in total cell protein (p < 0.01; Figure 2E) and a 12% higher protein to DNA ratio (proxy of hypertrophy) at 18 hours (p < 0.05) and 46% at 48 hours (p < 0.001). We also calculated the protein concentration:cells/hpf ratio of strained vs. control (56.4 ± 4.0 μg protein/cell vs. 28.8 ± 2.8; μg protein/cell p < 0.05) and in this case we found there to be a significant difference.
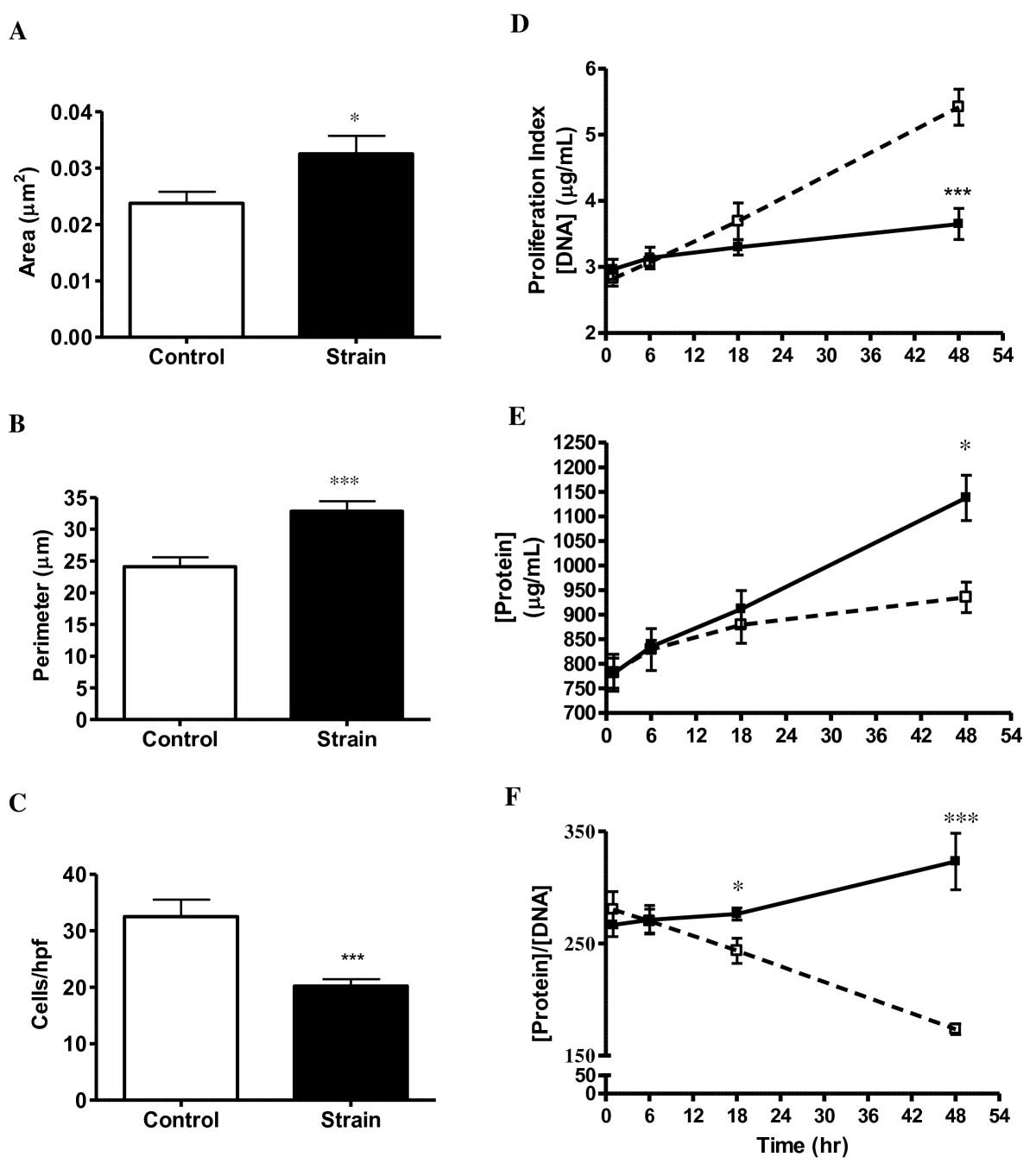
Figure 2
Figure 2 caption
Strain Regulation of VSMC Hyperplasia and Hypertrophy: VSMC area (A), perimeter (B), and cells per high power field (hpf; panel C) data from control (N = 17) and strained (N = 16) cells. Proliferation index (D), cellular protein (E), and protein/DNA ratio (F) data for control (open symbols) and strained (closed symbols) VSMC. N = 6 to 9 per group; *p < 0.05; ***p < 0.001 vs. control at the same time point.
Compared to control cells at 48 hours, strained VSMC exhibited significant changes in the secretion of CINC-3, CNTF, Fractalkine, GM-CSF, IL-1β, IL-4, IL-6, Leptin, MCP-1, β-NGF and VEGF (p < 0.05 for all; Figure 3). Further analysis of VEGF secretion via ELISA showed a 57% (p < 0.05) increase after 18 hours strain and 22% increase (p > 0.05) after 48 hours strain when compared to their respective controls (Figure 4A). VSMC also secreted 65% more NO after 48 hours of CS compared to control (p < 0.05; Table 1).
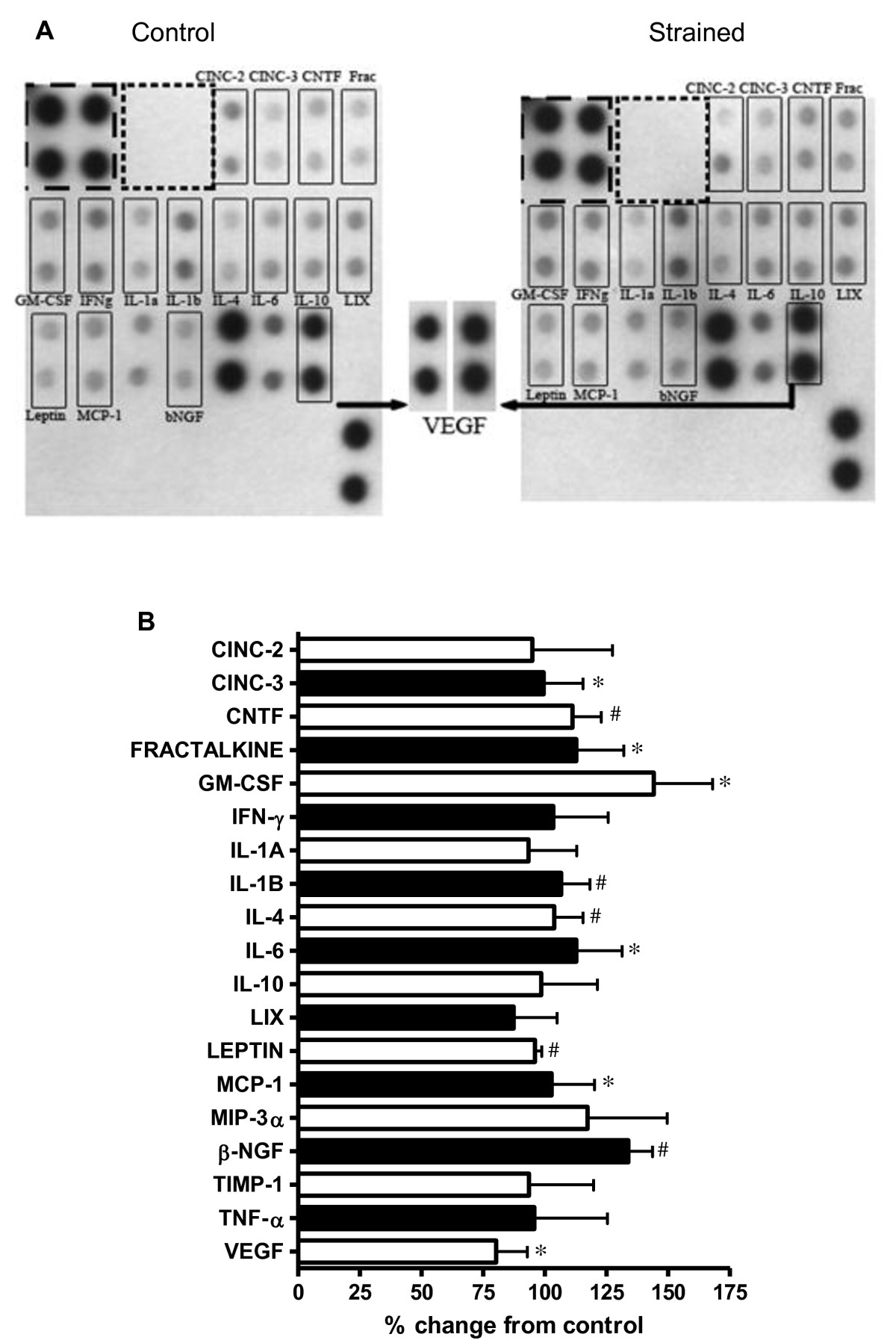
Figure 3
Figure 3 caption
Strain Regulation of VSMC Cytokine Secretion: Changes in cytokine levels in conditioned media from control and strained VSMC. (A) Representative matched protein microarrays from control (left) and strained (right) groups. VEGF signals have been enlarged for clarity. (B) Summary data for six protein microarrays showing percent change from control in cytokine levels *p < 0.05 vs. %CFC > 25%; p < 0.05 vs. %CFC > 50%. All data are corrected on a per-cell basis. (.
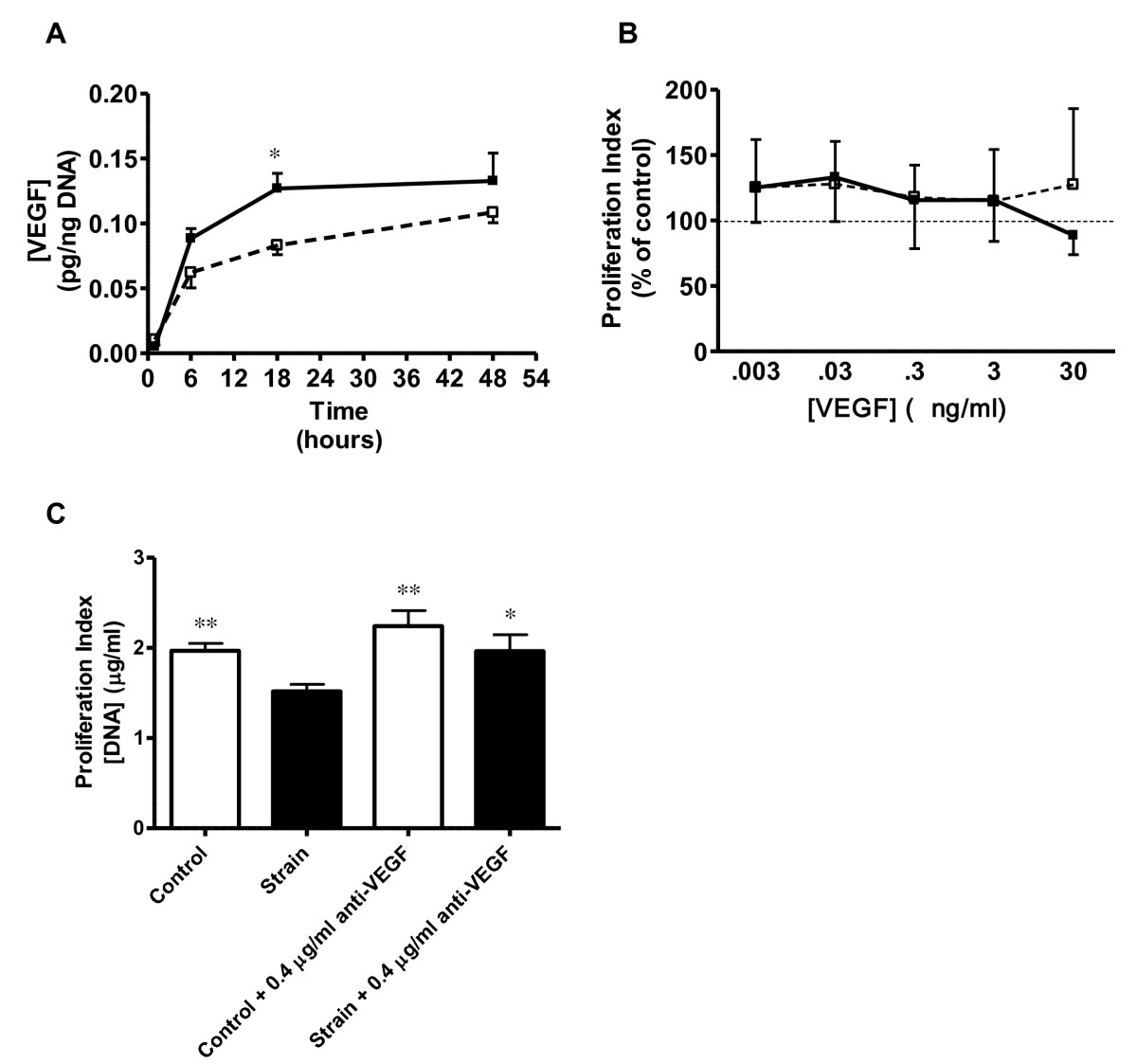
Figure 4
Figure 4 caption
Effects of Strain on VEGF Secretion and VEGF Blockade on VSMC Proliferation: (A) Temporal changes in VEGF secretion from control (open symbols) and strained VSMC. N = 5 per group; * p ≤ 0.05 vs. control at same time point. (B) Lack of significant effects of 18 hours (closed symbols) and 48 hours (open symbols) of exogenous VEGF treatment on proliferation in unstrained VSMC (N = 9). (C) Effects of strain (closed bars) and anti-VEGF treatment on VSMC proliferation. N = 6; *p < 0.05 vs strain only group; **p < 0.01 vs strain only group.
In the absence of strain, 18 and 48 hours incubation with various concentrations of exogenous VEGF did not affect VSMC proliferation when compared to control (Figure 4B). However, the addition of 0.4 μg/ml anti-VEGF (100 times neutralizing dose, 50%) to VSMC prior to a CS regimen caused a 29% increase (p < 0.01) in proliferation accompanied by 26% decrease in protein expression (p < 0.05) compared to strained VSMC without anti-VEGF (Figure 4C). Lower concentrations of anti-VEGF (0.004 and 0.04 ug/ml) were tested, but did not result in significant effects (results not shown). Importantly, anti-VEGF also did not significantly affect proliferation or protein expression in non-strained control cells.
There were no differences in proliferation among VSMC incubated with extracellular calcium, with calcium plus the SACC inhibitor GdCl3, or in the absence of extracellular calcium (osmotically controlled with Na+) after 48 hours strain. When compared to non-strain VSMC, proliferation among all strain groups were significantly decreased (Table 1).
Antibody protein microarrays of VSMC lysates revealed 46 cellular proteins and/or protein phosphorylation states that changed ± 25% from control (Table 2). Of interest, MAPK cascade peptides including MEK1, MEK2, phospo-MEK1T385, T291, S298, and phospho-Erk1/2T202+Y204/T185+T187 were universally increased in strained VSMC compared to control. PKCλ/τ, phospo-PKCλ/τT555, PKCζ, PKCθ, phospo-PKCθS695, PKCμ, phospo-PKCηT655, PKCε, phospo- PKCδS645 were also universally elevated compared to control. However, Cdk4, Cdk6, phospo-Cdk1/2T14+T15, and Cdk2 were all decreased in cell lysates of strained versus control (Table 2).
Table 2
Effects of strain on VSMC intracellular signaling.
| MEK1/ERK | PKC | CDK | |||
|---|---|---|---|---|---|
| MEK1 | +148% | PKC λ/ι | +41.4% | Cdk4 | -26% |
| MEK1T385 | +68.1% | PKC λ/ιT555 | +90.3% | Cdk6 | -39.3% |
| MEK1T291 | +159% | PKC ζ | +87% | Cdk1/2T14+Y15 | -50% |
| MEK1S298 | +62% | PKC θ | +231% | Cdk2 | -49.6% |
| MEK2 | +188.1% | PKC θS695 | +44.6% | ||
| ERKT202+Y204/T185+Y187 | +130% | PKC μ | +97.3% | ||
| PKC ε | +30% | ||||
| PKC δ | +40.5% | ||||
Discussion
Our study shows that cyclic strain (CS) causes an antimitogenic response in VSMC, changes to actin/cell alignment, hypertrophy, and strain-regulated cytokine secretion. Secretion of NO and VEGF, among other cytokines, are up-regulated in strained cells temporally corresponding to the observed decreases in proliferation. While exogenous VEGF alone (in the absence of strain) does not inhibit proliferation, neutralization of VEGF secreted from strained cells is effective in restoring normal proliferative responses in physiologically strained VSMC. These data suggest that VEGF is necessary, but is not sufficient to cause the antiproliferative effects seen with CS. The antimitogenic intracellular signaling mechanisms responsible are extracellular calcium-independent, but potentially mediated by downregulation of Cdks.
VSMC and their major actin stress fibers aligned nearly perpendicular to the dominant strain vector after 48 hours of CS, a response which is consistent with our previous findings [20]. In the
CS causes an antimitogenic response in VSMC's (Figure 2D and 4C). While we take into account VSMC in a uniculture-only model, such a response in vivo would cause leaky blood vessels with poor VSMC coverage [23]. In vivo, VSMC proliferation is likely influenced by other factors such as PDGF and IGF-1 released by endothelial cells [24] to maintain a growth equilibrium between strain induced anti-mitogenesis and paracrine mediated VSMC proliferation. While we recognize that other cell types can influence VSMC proliferation, for this study we were interested only in soluble mediators secreted by VSMC in response to strain that may self-regulate VSMC proliferation through an autocrine mediated manner.
Autocrine cytokines have been implicated in strain-induced hyperplasia (e.g., VEGF, IGF-1, PDGF, angiotensin II). We and others have found that some autocrines may be alternatively regulated in response to strain duration and magnitude. Quinn et al. has shown that a 15% equiradial strain increased the synthesis of VEGF mRNA, while a 25% equiradial strain had no affect VEGF mRNA [25]. This same group reported that while VEGF mRNA expression increases significantly after 24 hrs of strain, there was no difference when compared to non-strain cells after 48 hours strain [25]. We similarly observed a 1.8 fold increase in VEGF secretion at 18 hours which then leveled to control at 48 hours (Figure 4A). The addition of anti-VEGF polyclonal antibody was sufficient to reverse VSMC antimitogenic response to strain (Figure 4C) suggesting that VEGF is indeed a key mediator in the VEGF antiproliferative response. Others have shown in vitro exogenous VEGF (30 - 100 ng/ml) inhibits VSMC proliferation in static conditions [11, 26] possibly though inhibition of MEK1/2 - ERK1/2 phosphorylation. However, exogenous incubation of VEGF at concentrations approximate to secretory levels expressed in our model (0.03 ng/ml) was insufficient to inhibit proliferation in non-strain cultures (Figure 4B). This suggests that strain itself is an important cofactor in VEGF-induced antimitogenesis. Secretions of several other cytokines were upregulated in response to strain with documented roles in mediating both VSMC pro- and anti-mitogenic response; pro-inflammatory cytokines CINC-3, CNTF, Fractalkine, GM-CSF, IL-1β, IL-6, MCP-1, and BETA-NGF; anti-inflammatory cytokines IL-4 and IL-10; VEGF and leptin (Figure 3). The antiproliferative response we observed likely culminates from a balance among both pro- and anti-mitogenic cytokines working alongside VEGF. Exploring whether antimitogenesis is due to strain per se or resultant autocrine growth factors/cytokines in our experiments is to be determined.
Intracellular transmission of mechanical strain stimuli that induce VSMC mitogenic responses are highly dependent on the mitogen activated protein kinase (MAPK) cascade [1]. Mechanical strain is known to activate all three members of the MAP kinase cascade: ERK1/2, JNKs, p38 MAPK [27, 28]. Our results indicate that neither the JNK nor p38 MAPK were elevated during strain, suggesting these two signaling cascades were not activated by our strain paradigm (results not shown). However, our results did show ERK1/2 signaling cascade activation through MEK phosphorylation at threonines 385 and 291, and serine 298. These sites have been previously shown to induce MEK activation [29]. MEK1/2 then activate downstream ERK1/2 by phosphorylating ERK threonines 202 and 185 and ERK tyrosines 204 and 187 [30–32]. This is consistent with our findings of increased ERK1/2 phosphorylation at these sites (Table 2). Furthermore, protein kinase C (PKC) is also a key upstream regulator of ERK1/2 [33] which was also found to be upregulated in response to strain. This study shows 48 hours of CS activates PKC θ and δ by phosphorylation at serine 695 and serine 645 respectively and activates [34] atypical PKC λ/ι by phosphorylation of threonine 555 (Table 1). Although MAPK and PKC play important roles in strain-induced VSMC proliferation, the outcome of these signaling cascades, and thus progression through the cell cycle, is ultimately regulated by cyclin-Cdk complexes inside the cell nucleus which may be regulated in part by NO.
NO is a key mediator of the antimitogenic response which down regulates the expression of several Cdks [35]. Our previous studies have shown that NO is activated through the inducible nitric oxide synthase (iNOS) pathway during CS in VSMC cultured without serum [20]. Our latest results show NO production is significantly elevated vs. control (71%) after 48 hours of strain in the presence of serum (Table 1). This suggests that NO increases are strain-dependent and serum factor-independent. While our observed decrease in VSMC proliferation appears to be partially attributed to VEGF (Figure 4C), co-regulation by iNOS may also play a central role in strain-induced anti-proliferative response of VSMC.
Proliferation requires the expression of cyclin dependent kinases (Cdks) and our results show decreased expression of Cdk2, Cdk4, and Cdk6 after 48 hours of strain (Table 2). Decreased proliferation due to decreased expression of Cdk has been identified for VSMC suggesting that strain may be acting through a Cdk inhibitory pathway. NO-mediated downregulation of Cdk2 activity and cyclin A gene transcription [35] may help to explain our results which show a decrease in strain-induced VSMC proliferation despite MAPK and ERK1/2 upregulation. Decreased expression of both Cdk4 and Cdk6 also suggests attenuated numbers of cyclin D/Cdk4, Cdk6 complexes, and hence a slowing of VSMC movement past the G0/1 restriction point. Our results also showed a decrease in the phosphorylation of Cdk1/2 at threonine 14 and tyrosine 15 (Table 2). These two phosphorylation domains are critical for the inhibition of cyclin/Cdk complexes [36] suggesting a lack of proliferative inhibition via this mechanism. However, the down-regulation of Cdks 2, 4, and 6 in response to strain appears to override the mitogenic effect of decreased phosphorylation of Cdk1/2 resulting in a decrease in VSMC proliferation.
Conflict of interests
The authors declare that they have no competing interests.
Acknowledgements
The authors would like to acknowledge Dr. Kathryn Leyva and Dr. David Gardner for their thoughtful insight and input.
Sources of funding
This work was supported by the American Heart Association National Center, Midwestern University, and the Arizona Biomedical Research Collaborative.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
Authors’ original file for figure 2
Authors’ original file for figure 3
Authors’ original file for figure 4
References
- Biomechanical stress-induced signaling in smooth muscle cells: an update. Curr Vasc Pharmacol. 2003;1(1):41-58.
- Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J Biomech. 2007;40(5):947-60.
- Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15(3):100-8.
- Mechanical stretching stimulates smooth muscle cell growth, nuclear protein import, and nuclear pore expression through mitogen-activated protein kinase activation. J Biol Chem. 2007;282(32):23081-8.
- Expression of smooth muscle and nonmuscle myosin heavy chains in cultured vascular smooth muscle cells. J Biol Chem. 1986;261(31):14740-5.
- Induction of SM-alpha-actin expression by mechanical strain in adult vascular smooth muscle cells is mediated through activation of JNK and p38 MAP kinase. Biochem Biophys Res Commun. 2003;301(4):1116-21.
- Mechanical influences on vascular smooth muscle cell function. J Hypertens. 1998;16(12 Pt 2):1921-9.
- Cyclic stretch regulates autocrine IGF-I in vascular smooth muscle cells: implications in vascular hyperplasia. Am J Physiol. 1999;276(4 Pt 1):E697-705.
- Activation of mitogenic and antimitogenic pathways in cyclically stretched arterial smooth muscle. Am J Physiol Endocrinol Metab. 2001;281(6):E1165-71.
- Mechanical strain induces growth of vascular smooth muscle cells via autocrine action of PDGF. J Cell Biol. 1993;123(3):741-7.
- Vascular endothelial growth factor inhibits mitogen-induced vascular smooth muscle cell proliferation. J Surg Res. 2003;114(2):179-86.
- Dual cell cycle-specific mechanisms mediate the antimitogenic effects of nitric oxide in vascular smooth muscle cells. J Hypertens. 1997;15(3):275-83.
- Hypoxia-inducible factor-1alpha/vascular endothelial growth factor pathway for adventitial vasa vasorum formation in hypertensive rat aorta. Hypertension. 2002;39(1):46-50.
- Cyclic stretch induces the expression of vascular endothelial growth factor in vascular smooth muscle cells. Endothelium. 2001;8(1):41-8.
- Mechanical stress-initiated signal transductions in vascular smooth muscle cells. Cell Signal. 2000;12(7):435-45.
- Strain profiles for circular cell culture plates containing flexible surfaces employed to mechanically deform cells in vitro. J Biomech. 1994;27(9):1169-77.
- In vitro biophysical strain model for understanding mechanisms of osteopathic manipulative treatment. J Am Osteopath Assoc. 2006;106(3):157-66.
- Modeled repetitive motion strain and indirect osteopathic manipulative techniques in regulation of human fibroblast proliferation and interleukin secretion. J Am Osteopath Assoc. 2007;107(12):527-36.
- Biostatistical Analysis 4th Edition. Prentice Hall. 1998.
- Cyclic stretch induces vascular smooth muscle cell alignment via NO signaling. Am J Physiol Heart Circ Physiol. 2002;283(5):H1907-14.
- Leptin induces vascular smooth muscle cell hypertrophy through angiotensin II- and endothelin-1-dependent mechanisms and mediates stretch-induced hypertrophy. J Pharmacol Exp Ther. 2005;315(3):1075-84.
- Differential effects of pressure and flow on DNA and protein synthesis and on fibronectin expression by arteries in a novel organ culture system. Circ Res. 1995;77(4):684-94.
- A role for VEGF as a negative regulator of pericyte function and vessel maturation. Nature. 2008;456(7223):809-13.
- Influence of endothelial cells on vascular smooth muscle cells phenotype after irradiation: implication in radiation-induced vascular damages. Am J Pathol. 2006;169(4):1484-95.
- Cyclic mechanical stretch induces VEGF and FGF-2 expression in pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L897-903.
- Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol. 2001;188(3):359-68.
- Cyclic strain stress-induced mitogen-activated protein kinase (MAPK) phosphatase 1 expression in vascular smooth muscle cells is regulated by Ras/Rac-MAPK pathways. J Biol Chem. 1999;274(36):25273-80.
- Mechanical stress-induced heat shock protein 70 expression in vascular smooth muscle cells is regulated by Rac and Ras small G proteins but not mitogen-activated protein kinases. Circ Res. 2000;86(11):1122-8.
- The threonine residues in MAP kinase kinase 1 phosphorylated by MAP kinase in vitro are also phosphorylated in nerve growth factor-stimulated rat phaeochromocytoma (PC12) cells. FEBS Lett. 1994;341(1):119-24.
- Purification and properties of extracellular signal-regulated kinase 1, an insulin-stimulated microtubule-associated protein 2 kinase. Biochemistry. 1991;30(1):278-86.
- ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65(4):663-75.
- Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). Embo J. 1991;10(4):885-92.
- Strain activation of bovine aortic smooth muscle cell proliferation and alignment: study of strain dependency and the role of protein kinase A and C signaling pathways. J Cell Physiol. 1997;170(3):228-34.
- Crystal structure of the catalytic domain of human atypical protein kinase C-iota reveals interaction mode of phosphorylation site in turn motif. J Mol Biol. 2005;352(4):918-31.
- Nitric oxide-induced downregulation of Cdk2 activity and cyclin A gene transcription in vascular smooth muscle cells. Circulation. 1998;97(20):2066-72.
- Cyclins and cyclin-dependent kinases: a biochemical view. Biochem J. 1995;308(Pt 3):697-711.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.