Erratum to: The importance of microglia in the development of the vasculature in the central nervous system
Vascular Cell. 2013;
Received: 10 June 2013 | Accepted: 10 June 2013 | Published: 25 June 2013
Vascular Cell ISSN: 2045-824X
Correction
After the publication of this work [1] it was brought to our attention that citations in the article were not correspondingly numbered in the reference list. To avoid confusion, the article is republished here in its entirety, with the citations referenced correctly.
The Publisher and authors apologize to the readers for the inconvenience caused.
Abstract
The body’s vascular system is thought to have developed in order to supply oxygen and nutrients to cells beyond the reach of simple diffusion. Hence, relative hypoxia in the growing central nervous system (CNS) is a major driving force for the ingression and refinement of the complex vascular bed that serves it. However, even before the establishment of this CNS vascular system, CNS-specific macrophages (microglia) migrate into the brain. Recent studies in mice point to the fundamental importance of microglia in shaping CNS vasculature during development, and re-shaping these vessels during pathological insults. In this review, we discuss the origin of CNS microglia and their localization within the brain based on data obtained in mice. We then review evidence supporting a functional role of these microglia in developmental angiogenesis. Although pathologic processes such as CNS ischemia may subvert the developmental functions of microglia/macrophages with significant effects on brain neo-angiogenesis, we have left this topic to other recent reviews [2, 3].
Microglia – specialized macrophages of the CNS
Microglia are specialized macrophages of the central nervous system involved in immune regulation, tissue development, homeostasis and wound repair. Microglia were first observed by Virchow in the mid-nineteenth century (see [4]), and described in greater detail by Pio del Rio-Hortega in 1932. In this almost prescient work, del Rio-Hortega described microglia morphology, plasticity during development and with pathological insult, their cellular origin, and microglia association with white matter tracts and blood vessels. Despite an immense amount of research on microglia origin and function since then, these early views remain surprisingly accurate.
Microglia derive from primitive yolk sac macrophages
Microglia belong to the mononuclear phagocytic system - a family of cells that includes committed precursors in the bone marrow, circulating blood monocytes and tissue macrophages in every organ of the body including the CNS [5]. Mononuclear phagocytes are typified by their ability to ingest large particles; their morphology; their expression of common surface markers including CD11b, CD68, Colony Stimulating Factor 1 Receptor (CSF1R), chemokine receptor CXCR3, and plasma membrane glycoprotein F4/80 [6]; and their presumed hematopoietic origin [5]. While microglia certainly meet the functional and morphological definition of a mononuclear phagocyte [7–9], their developmental origin has until recently been less clear.
In mice, hematopoietic stem cells (HSCs) emerge from the dorsal aorto-gonado-mesonephros (AGM) region 10.5 days after conception (embryonic day (E) 10.5), then migrate to the fetal liver where they expand and differentiate before definitive hematopoiesis in the spleen and bone marrow [10–13]. In adult mice, blood monocytes, classical dendritic cells, and certain tissue macrophages derive from, and are continuously replaced by, bone marrow-derived HSCs. It was previously thought that microglia arose from hematopoietic precursors in two waves of recruitment and differentiation [14, 15]. However, it is now clear, based on evidence from bird, fish and mammals, that yolk-sac derived macrophage precursors contribute significantly, if not entirely, to the brain’s microglia. In avian embryos, analyses using chick-quail transplantation and parabiosis chimeras show that yolk sac-derived macrophages migrate to and invade the CNS through the pial basal lamina before and independent of CNS vacularization [16, 17]. Subsequent live recordings of cell movements in zebrafish embryos revealed that yolk sac-derived macrophages migrate through the cephalic mesenchyme before its vascularization to reach the brain pial surface and the roof of the 4th ventricle, from where they subsequently invade the neuroepithelium and eventually acquire microglial characteristics [18]. Recently, fate mapping studies in the mouse using genetic lines such as
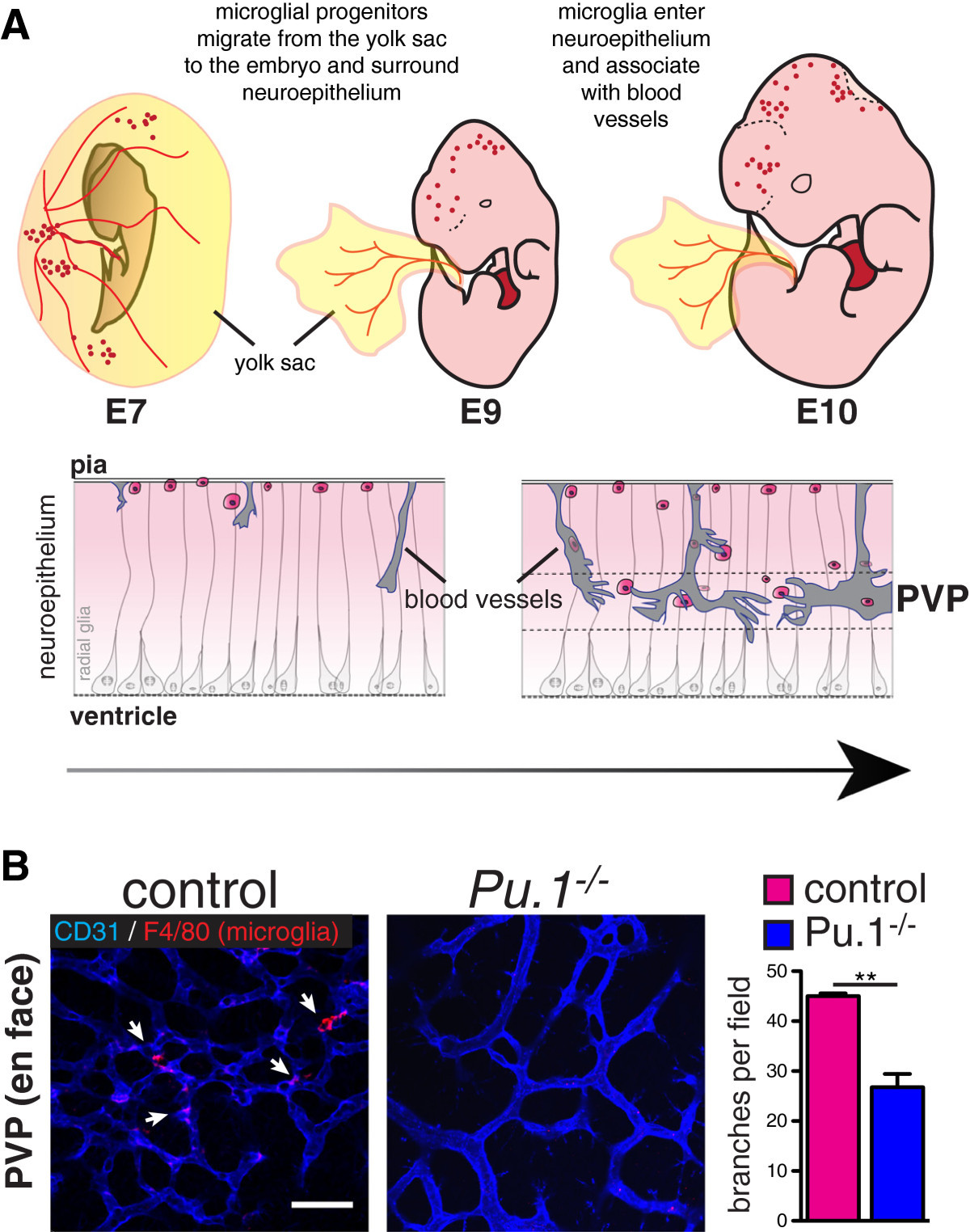
Figure 1
Figure 1 caption
A) Microglia originate from myeloid precursors in the yolk sac, which migrate into the neuroepithelium by E10. They associate with radial glia and with blood vessels (also ingressing into the brain from the pial surface) where they may promote fusion of vascular tip cells in the periventricular vascular plexus (PVP). Arrow indicates progressive development from mouse embryonic day (E) 7 to E10. (Modified from [].) B) Bottom: Reduced vascular branching in the brains of microglia-deficient mouse embryos. Flat mounted brains from embryonic day 12 mice stained for endothelium (CD31, blue) and microglia (F4/80, red). Branch points quantified from 4 mutants and controls. Students T Test P < 0.005.
The yolk sac origin of microglia is further supported by elegant experiments done in mice lacking the transcription factors PU.1 [20, 25–27] or Myb [20], and in mice with inactivated
Patterns of brain colonization by microglia
Once born, yolk-sac derived macrophage precursors migrate into and colonize the whole (mouse) embryo between E9.5 and E10.5 (Figure 1) [19, 20]. The first organ to be colonized is the brain. Subsequent phases of microglial brain colonization follow a stereotyped pattern (see [33]). Microglia invade the brain through the pial surface, then migrate and proliferate, populating the brain in a dorsal-to-ventral and rostral-to-caudal gradient. During this time, microglia associate with radial glia and blood vessels, and are found in close proximity to dying cells. Eventually, microglia are notably excluded from the neuroepithelium and cortical plate, and then are widely distributed in the adult brain, except in areas of densely packed neuron cell bodies such as the pyramidal cell layer. The early association of microglia around blood vessels has led to the hypothesis that microglia may enter the brain through the developing vasculature. Indeed, E10.5
Microglia shape the developing CNS vasculature
Because the developing CNS lacks intrinsic vasculature, CNS blood vessel development occurs exclusively via angiogenesis [39–41]. Attracted by proangiogenic signals, new capillaries sprout from perineural vessels, and invade the neuroectoderm around E10 in mice. These nascent capillaries are composed of tip cells at the vascular front, followed by proliferative stalk cells. Tip cells extend filopodia toward guidance cues such as VEGF-A. VEGF-A induces expression of the Notch ligand, Dll4, predominately in tip cells. Dll4 then activates Notch in adjacent cells, which down-regulates VEGF receptors and up-regulates angio-suppressive factors like sFlt1 and Jagged-1, promoting a stalk cell phenotype (reviewed in [41]). The interplay between VEGF and Notch signaling is highly regulated with additional inputs from other major signaling pathways including BMPs [42, 43], Semaphorins [44], and Wnt/βcatenin [45, 46]. Additional signaling pathways that regulate tip cell formation and sprouting include sphingosine-1-phosphate and its receptor S1pr1 [47–49]. During vascular sprouting, tip cells anastomose with neighboring tip cells, creating vascular loops. In this way, vessels sprout, extend, branch and anastomose, iteratively, toward the center of the neural tube where they establish a temporary plexus, termed the periventricular vascular plexus (PVP), around the CNS ventricular spaces and spinal cord’s central canal [50–52]. As the CNS grows and differentiates, these vessels associate with microglia, pericytes, neuroepithelial radial glia and neuroblasts, and later astrocytes; CNS vessels are refined, arteries and veins are established, and the mature neurovascular system takes form.
The retina and optic nerve represent highly specialized extensions of the forebrain. While its vascularization occurs by angiogenic sprouting similar to the brain, the timing and scaffolds that guide angiogenesis are partly different [53]. During the first week of life, an astrocytic network arising from the optic nerve invades into the retina in a centrifugal fashion. As the primitive hyaloid (hv) vessels that supported the embryonic eye development regress, a new primary vascular plexus extends into the retina, following structural and morphogenic cues provided by astrocytes and Müller glia. At 7 to 9 days of postnatal life, vessels sprout perpendicularly into deeper layers of the retina, forming a deep vascular plexus in the outer plexiform retinal layer (OPL). During the next 3 weeks, retinal vasculature continues to sprout, remodel and differentiate into arteries and veins and a mature neurovascular network is established by 6 weeks of age.
Microglia influence CNS vascular development
As discussed above, microglia migrate into the CNS and retinal neuroepithelium before vessels do. Microglia are therefore uniquely positioned to influence the early sprouting, migration, anastomosis, and refinement of the growing CNS and retinal vascular systems. Studies of angiogenesis after microglial depletion, or in mice lacking microglia, strongly support this concept. Checchin et al. [54] administered clodronate liposomes either systemically to deplete macrophages and circulating monocytes, or intravitreally, which depleted retinal microglia without reducing circulating monocytes. These authors found that reducing retinal microglia numbers was associated with a decrease in retinal vascular density. Selective depletion of circulating monocytes had no impact on retinal blood vessels. Importantly, intravitreal co-administration of microglia with clodronate restored vascularity of the developing retina, suggesting a microglia-specific effect on retinal vascular development. Similarly, Kubota et al. [38] found that
The concept that microglia may act to “bridge” vascular sprouts during CNS vascular development was introduced by the work of Fantin et al. [21]. This group studied vascular ingression and branching in embryonic hindbrains and retinas of mice lacking macrophages and microglia (
A recent report from our laboratory confirmed and extended many of these observations [37]. We found that microglia-deficient mice (
In contrast to these findings, Stefater et al. [56] recently uncovered a mechanism whereby microglia may
The feedback loop between VEGF and Notch involves regulation of both VEGFR-2 and VEGFR-3, although the individual contribution of each of these VEGF receptors remains unclear [40, 57–59]. The primary ligands for VEGFR-3, VEGF-C and VEGF-D, are highly expressed by microglia, and VEGF-C-positive microglia are found near the fusion points of VEGFR-3-positive vascular sprouts [59]. While
Other groups have recently explored potential roles for Notch signaling in microglia-endothelial cell interactions. Outtz et al. [60] found that Notch signaling is activated in retinal microglia, which are closely associated with endothelial tip cells expressing the Notch ligand Dll4. Moreover, genetic deletion of Notch1 in retinal microglia led to a subtle reduction in the numbers of microglia found at the vascular front. Interestingly, Hoffman et al. [61] found that the Notch ligand, Jagged1, is highly expressed in perivascular cells of the retina, including microglia. Further studies should evaluate the specific roles of Notch signaling in microglia, and the impact of this signaling on retinal vascular development.
Outlook
The development of organ-specific vascular beds is dependent upon the close communication between the vascular cells (endothelial cells and mural cells) on the one hand, and resident cells of the organ, on the other. The CNS vasculature is in many ways unique in its anatomy and regulation, and it harbors a highly specific barrier – the blood–brain, or blood-retina, barrier. The development of the CNS vasculature hence occurs in tight association with the development of other components of the CNS, and as a result of reciprocal communication between the endothelial cells and different emerging CNS cell types. This review focuses on the role of microglial cells – a CNS-specific type of macrophage – and also mentions in passing the importance of other cells types, such as radial glial cells and astrocytes, as sources of VEGFs, Wnts and other signaling molecules that control the shape and function of the emerging vasculature. Although recent studies provide compelling evidence for a role of microglia in shaping the nascent vascular plexuses in the brain and retina, the molecular mechanisms remain to be elucidated, and the possibility remains that microglia play a different role at different locations, for example in the different retinal capillary plexuses. It also remains to be shown what role, if any, microglia play in vascular homoeostasis in adult physiological and pathophysiological processes. Microglial cells become activated in conjunction with pathological insults and disruption of the blood–brain barrier, where they may play protective or pathogenic roles. These data are not discussed in the present review, which is focused on physiological development. However, the awareness of microglia as a unique CNS cell type with a distinct ontogeny and equipped with specific developmental and pathological functions compared to other glial cell types, has recently increased. Our perspectives on these cells will undoubtedly grow rapidly in the coming years.
Ethical approval
Animal experiments were approved by the Stockholm’s North Ethical Committee for Animal Research.
Acknowledgments
This work has been supported by a Leducq Foundation Transatlantic Network of Excellence and a Transatlantic Career Development Award.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Authors’ original file for figure 1
{kind=link}
References
- The importance of microglia in the development of the vasculature in the central nervous system. Vascular Cell. 2013;5:4-.
- Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9:259-270.
- Metchnikoff's policemen: macrophages in development, homeostasis and regeneration. Trends Mol Med. 2011;17:743-752.
- Physiology of microglia. Physiol Rev. 2011;91:461-553.
- The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull World Health Organ. 1972;46:845-.
- Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723-737.
- Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387-1394.
- Live imaging of neuronal degradation by microglia reveals a role for v0-atpase a1 in phagosomal fusion in vivo. Cell. 2008;133:916-927.
- Long-range ca2+ waves transmit brain-damage signals to microglia. Dev Cell. 2012;22:1138-1148.
- Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature. 2007;446:1056-1061.
- Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464:112-115.
- In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010;464:116-120.
- Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464:108-111.
- Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience. 1985;15:313-326.
- The origin and cell lineage of microglia: New concepts. Brain Res Rev. 2007;53:344-354.
- Embryonic CNS macrophages and microglia do not stem from circulating, but from extravascular precursors. GLIA. 1998;22:98-102.
- First appearance, distribution, and origin of macrophages in the early development of the avian central nervous system. J Comp Neurol. 1993;330:113-129.
- Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive process. Dev Biol. 2001;238:274-288.
- Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841-845.
- Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K: A lineage of myeloid cells independent of myb and hematopoietic stem cells. Science. 2012;336:86-90.
- Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of vegf-mediated endothelial tip cell induction. Blood. 2010;116(5):829-840.
- Expression of gal4-dependent transgenes in cells of the mononuclear phagocyte system labeled with enhanced cyan fluorescent protein using csf1r-gal4vp16/UAS-ECFP double-transgenic mice. J Leukoc Biol. 2008;83:430-433.
- Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. 2011;14:1142-1149.
- Microglia and monocytes: Tis plain the twain meet in the brain. Nat Neurosci. 2011;14:1098-1100.
- PU.1 regulates the commitment of adult hematopoietic progenitors and restricts granulopoiesis. J Exp Med. 2005;201:1487-1502.
- Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647-5658.
- PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998;12:2403-2412.
- Microglia in colony-stimulating factor 1-deficient op/op mice. J Neurosci Res. 1995;40:535-544.
- Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6:e26317-.
- Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39-48.
- Opposing actions of c-ets/PU.1 and c-myb protooncogene products in regulating the macrophage-specific promoters of the human and mouse colony-stimulating factor-1 receptor (c-fms) genes. J Exp Med. 1994;180:2309-2319.
- Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. 2010;88:495-505.
- Physiological roles of microglia during development. J Neurochem. 2011;119:901-908.
- Targeted inactivation of the sodium-calcium exchanger (ncx1) results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J. 2001;15:1209-1211.
- Pattern of invasion of the embryonic mouse spinal cord by microglial cells at the time of the onset of functional neuronal networks. GLIA. 2011;59:675-695.
- Embryonic and postnatal development of microglial cells in the mouse retina. J Comp Neurol. 2008;506:224-239.
- A two-way communication between microglial cells and angiogenic sprouts regulates angiogenesis in aortic ring cultures. PLoS One. 2011;6:e15846-.
- M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 2009;206:1089-1102.
- Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol. 2010;22:617-625.
- VEGF and notch in tip and stalk cell selection. Cold Spring Harb Perspect Med. 2013;3(1):a006569-.
- Angiogenesis: A team effort coordinated by notch. Dev Cell. 2009;16:196-208.
- Stalk cell phenotype depends on integration of notch and smad1/5 signaling cascades. Dev Cell. 2012;22:501-514.
- ALK1 signaling inhibits angiogenesis by cooperating with the notch pathway. Dev Cell. 2012;22:489-500.
- Semaphorin 3e–plexin-d1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 2011;25:1399-1411.
- The wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating dll4/notch signaling. Dev Cell. 2010;18:938-949.
- Rspo1/wnt signaling promotes angiogenesis via vegfc/vegfr3. Development. 2011;138:4875-4886.
- S1P1 inhibits sprouting angiogenesis during vascular development. Development. 2012;139:3859-3869.
- Flow-regulated endothelial S1P receptor-1 signaling sustains vascular development. Dev Cell. 2012;23:600-610.
- The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between ve-cadherin and VEGFR2. Dev Cell. 2012;23:587-599.
- Compartment-specific transcription factors orchestrate angiogenesis gradients in the embryonic brain. Nat Neurosci. 2008;11:429-439.
- Vascular architecture of the developing spinal cord in the rat: A suggested model. J Embryol Exp Morphol. 1983;76:27-36.
- From angiogenesis to neuropathology. Nature. 2005;438:954-959.
- Development of the retinal vasculature. Angiogenesis. 2007;10:77-88.
- Potential role of microglia in retinal blood vessel formation. Invest Ophthalmol Vis Sci. 2006;47:3595-3602.
- SDF-1/CXCR4 contributes to the activation of tip cells and microglia in retinal angiogenesis. Invest Ophthalmol Vis Sci. 2010;51:3362-3371.
- Regulation of angiogenesis by a non-canonical wnt-flt1 pathway in myeloid cells. Nature. 2011;474:511-515.
- Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature. 2012;484:110-114.
- Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454:656-660.
- VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing notch signalling. Nat Cell Biol. 2011;13:1202-1213.
- Notch1 controls macrophage recruitment and notch signaling is activated at sites of endothelial cell anastomosis during retinal angiogenesis in mice. Blood. 2011;118:3436-3439.
- Notch expression patterns in the retina: An eye on receptor-ligand distribution during angiogenesis. Gene Expr Patterns. 2007;7:461-470.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.