γ-Actin plays a key role in endothelial cell motility and neovessel maintenance
Vascular Cell. 2015;
Received: 17 September 2014 | Accepted: 29 December 2014 | Published: 6 February 2015
Vascular Cell ISSN: 2045-824X
Abstract
Background
Angiogenesis plays a crucial role in development, wound healing as well as tumour growth and metastasis. Although the general implication of the cytoskeleton in angiogenesis has been partially unravelled, little is known about the specific role of actin isoforms in this process. Herein, we aimed at deciphering the function of γ-actin in angiogenesis.
Methods
Localization of β- and γ-actin in vascular endothelial cells was investigated by co-immunofluorescence staining using monoclonal antibodies, followed by the functional analysis of γ-actin using siRNA. The impact of γ-actin knockdown on the random motility and morphological differentiation of endothelial cells into vascular networks was investigated by timelapse videomicroscopy while the effect on chemotaxis was assessed using modified Boyden chambers. The implication of VE-cadherin, VEGFR-2 and ROCK signalling was then examined by Western blotting and using pharmacological inhibitors.
Results
The two main cytoplasmic isoforms of actin strongly co-localized in vascular endothelial cells, albeit with some degree of spatial preference. While β-actin knockdown was not achievable without major cytotoxicity, γ-actin knockdown did not alter the viability of endothelial cells. Timelapse videomicroscopy experiments revealed that γ-actin knockdown cells were able to initiate morphological differentiation into capillary-like tubes but were unable to maintain these structures, which rapidly regressed. This vascular regression was associated with altered regulation of VE-cadherin expression. Interestingly, knocking down γ-actin expression had no effect on endothelial cell adhesion to various substrates but significantly decreased their motility and migration. This anti-migratory effect was associated with an accumulation of thick actin stress fibres, large focal adhesions and increased phosphorylation of myosin regulatory light chain, suggesting activation of the ROCK signalling pathway. Incubation with ROCK inhibitors, H-1152 and Y-27632, completely rescued the motility phenotype induced by γ-actin knockdown but only partially restored the angiogenic potential of endothelial cells.
Conclusions
Our study thus demonstrates for the first time that β-actin is essential for endothelial cell survival and γ-actin plays a crucial role in angiogenesis, through both ROCK-dependent and -independent mechanisms. This provides new insights into the role of the actin cytoskeleton in angiogenesis and may open new therapeutic avenues for the treatment of angiogenesis-related disorders.
Keywords
Cytoskeleton Actin Angiogenesis Vascular endothelial cells ROCK signallingBackground
Angiogenesis is defined as the formation of new blood vessels from pre-existing ones. It is crucial for organ growth during development but also throughout adult life to repair wounded tissues. Furthermore, an imbalance in this process directly contributes to numerous pathologies such as cancer, diabetes, age-related macular degeneration, ischemic disorders and rheumatoid arthritis [1,2]. The multi-step and complex process leading to the formation of a new vascular network relies on the activation of endothelial cells followed by their proliferation, migration and morphological differentiation into capillary tubes. The cytoskeleton which directly regulates and controls an impressive array of cell functions, including cell shape maintenance, cell division, vesicle and organelle transport, cell motility and differentiation, plays a major role in angiogenesis. Studies focusing on the anti-angiogenic properties of microtubule-targeting drugs – reviewed by Pasquier et al. [3] – have provided major insights into the role of microtubules in this process. However, very little is known about the specific role of actin isoforms in angiogenesis.
In vertebrates, there are 6 functional actin genes and the expression of the six actin isoforms is regulated both spatially and temporally in a tissue-specific manner. Four of these isoforms (i.e. α-cardiac muscle actin, α-skeletal muscle actin, α-smooth muscle actin and γ-smooth muscle actin) are mainly expressed in muscle cells, while the cytoplasmic isoforms β- and γ-actin are ubiquitous [4]. Interestingly, the β- and γ-actin isoforms are almost identical proteins, differing only by 4 amino acid residues at the N-terminal end (positions 1, 2, 3 and 9). Distinct localization of β- and γ-actin mRNAs in several cell types, such as neurons, myoblasts and osteoblasts, has suggested for almost 20 years a spatial segregation of the two isoforms [5,6]. However, the spatial and functional segregation of β- and γ-actin was confirmed only recently in fibroblasts and epithelial cells by Chaponnier and colleagues, using newly developed monoclonal antibodies [7]. In particular, β-actin appears to play a role in cell attachment and contraction by preferentially localizing to stress fibres whereas γ-actin is mainly organised as a meshwork in cortical and lamellipodial structures and thus plays a crucial role in cell motility [7]. In accordance with this finding, we recently demonstrated that γ-actin specifically regulates cell motility by modulating the Rho-associated kinase (ROCK) signalling pathway and therefore influencing the phosphorylation of focal adhesion protein paxillin and myosin regulatory light chain 2 in neuroblastoma cells [8]. Elsewhere, key functional differences between β- and γ-actin were also recently revealed by mouse knock-out experiments. Indeed, β-actin knock-out mice are not viable, in part due to severe growth and migration defects of β-actin null embryonic cells, which are not observed in γ-actin null embryonic cells [9]. In contrast, γ-actin knock-out mice are viable, despite suffering increased mortality at birth and progressive hearing loss, which suggests that γ-actin is required for cytoskeleton maintenance but not for development [10]. Spatial segregation and functional differences led us to hypothesize that β- and γ-actin may play distinct roles in endothelial cells and differentially contribute to angiogenesis. We therefore investigated the localization of β- and γ-actin in vascular endothelial cells and undertook the functional analysis of γ-actin by RNAi to decipher its specific function in endothelial cell adhesion, motility and morphological differentiation into vascular networks, thus revealing a key role in angiogenesis.
Material and methods
Cell culture
HMEC-1 endothelial cells were originally isolated from dermal microvessels and immortalized by transfection with SV40 large T antigen [11]. They were obtained from the Cell Culture Laboratory in the Hôpital de la Conception (Assistance Publique Hôpitaux de Marseille, Marseille, France) and grown in MCDB-131 medium (Invitrogen, Mount Waverley, Australia) containing 10% heat-inactivated Fetal Calf Serum (FCS), 2 mM L-glutamine, 1% penicillin and streptomycin, 1 μg/mL hydrocortisone and 10 ng/mL epithelial growth factor (BioScientific, Gymea, Australia). BMH29L cells are bone marrow derived endothelial cells that were immortalized by ectopic expression of human telomerase reverse transcriptase [12]. They were kindly provided by Dr Karen MacKenzie (Children’s Cancer Institute Australia) and grown in Medium 199 (Invitrogen) containing 10% heat-inactivated FCS, 5% male human serum AB only (Sigma-Aldrich, Castle Hill, Australia), 1% penicillin and streptomycin, 1% heparin, 5 ng/mL recombinant human FGFβ (fibroblast growth factor β; Sigma-Aldrich) and 20 μg/mL Endothelial Cell Growth Factor (ECGF; Roche, Dee Why, Australia). Both cell lines were routinely maintained in culture on 0.1% gelatin-coated flasks at 37°C and 5% CO2. Cell lines were regularly screened and are free from mycoplasma contamination.
Gene silencing
γ-actin gene expression was silenced in endothelial cells using the siRNA sequence previously described (5′-AAGAGATCGCCGCGCTGGTCA-3′; Qiagen, Doncaster, Australia) [13]. An alternative siRNA sequence (5′-CAGCAACACGTCATTGTGTAA-3′; Qiagen) was also used in confirmation experiments [8]. β-actin gene expression was targeted using the siRNA sequence previously described (5′-AATGAAGATCAAGATCATTGC-3′; Qiagen) [14]. The optimum amount of siRNA was determined to be 200 and 500 pmol for HMEC-1 and BMH29L cells, respectively and was used in all subsequent experiments. A non-silencing control siRNA, which has no sequence homology to any known human gene sequence, was used as a negative control in all experiments (Qiagen). Cells were transfected using the Nucleofector® II device (Lonza, Mount Waverley, Australia) as previously described [15]. Briefly, HMEC-1 and BMH29L cells were resuspended in nucleofector® solution R and V, respectively, and transfected with siRNA using specifically optmized nucleofector® programs (T-016 and S-003 for HMEC-1 and BMH29L, respectively). All subsequent experiments were performed 72 h after siRNA transfection, when the level of γ-actin protein expression was the lowest.
Quantitative RT-PCR
The expression of γ-actin mRNA was examined using quantitative RT-PCR. Total RNA was extracted and DNase treated using the Qiagen RNeasy Plus kit according to the manufacturer’s instructions (Qiagen) and cDNA synthesis was performed using High capacity cDNA reverse transcription kit with RNAse inhibitor (Applied Biosystems, Mulgrave, Australia). Real-time PCR was performed on 7900HT Fast Real-time PCR system using the TaqMan® gene expression Master Mix (Applied Biosystems). γ-actin mRNA primer and probe sequences used were as follows: forward, 5′-CAGCTCTCGCACTCTGTTCTTC-3′; reverse, 5′-ACATGCCGGAGCCATTGT-3′; probe, 5′-CGCGCTGGTCATT-3′. All data were normalized to the housekeeping gene
Western blotting analysis
For western blotting analysis, cells were lysed in RIPA buffer containing a cocktail of protease and phosphatase inhibitors (Sigma-Aldrich). Equal amounts of protein (10–15 μg) were resolved on 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis or 4-15% pre-cast Criterion acrylamide gels (Bio-Rad Laboratories, Gladesville, Australia) before electrotransfer onto nitrocellulose membrane. Immunoblotting was performed using antibodies directed against β-actin (clone AC-74, Sigma-Aldrich), γ-actin – courtesy of Pr Peter Gunning [17], GAPDH (Abcam, Cambridge, UK), phospho-myosin light chain 2 (Cell Signaling Technology, Beverly, MA, USA), VE-cadherin (Cell signaling technology) and VEGFR-2 (Cell signaling technology). The membranes were then incubated with horseradish peroxidase-conjugated IgG secondary antibodies and protein detected with ECL Plus (GE Healthcare Life Sciences, Uppsala, Sweden). The blots were scanned and densitometric analysis performed as previously described [13].
Immunofluorescence staining
Vascular endothelial cells were seeded on gelatin-coated 8-well Permanox Lab-Tek chamber slides (Applied Biosystems) after siRNA transfection. β-actin and γ-actin were stained as previously described [7] with slight modifications. Specifically, cells were fixed with 3.7% formaldehyde for 20 min at RT and permeabilized with 100% methanol for 20 min at −20°C. Cells were incubated with the following primary mAbs: anti-β-actin (mAb 4C2, IgG1 – courtesy of Pr Christine Chaponnier [7]) and anti-γ-actin (mAb 2A3, IgG2b – courtesy of Pr Christine Chaponnier [7]). The following secondary Abs were used: FITC-conjugated goat anti-mouse IgG1 (Southern Biotechnology, Birmingham, AL) and TRITC-conjugated goat anti-mouse IgG2b (Southern Biotechnology). For tubulin staining, cells were fixed and permeabilized in 100% methanol at 20°C for 15 min and blocked with 10% FCS for 30 min. Microtubules were then stained with anti-βI-tubulin primary antibody (Abcam), followed by Alexa Fluor 488 anti-mouse secondary antibody (Invitrogen). For paxillin and phalloidin dual staining, cells were fixed with 3.7% formaldehyde/PBS for 10 min and permeabilized with 0.1% Triton X-100/PBS for 5 min. Focal adhesions were then stained with anti-paxillin primary antibody (BD Biosciences), followed by Alexa Fluor 488 anti-mouse secondary antibody (Invitrogen) and Alexa 568-conjugated phalloidin (Invitrogen). All slides were mounted on coverslips with ProLong Gold anti-fade reagent containing DAPI (Invitrogen) and imaged using the 63X oil-immersion objective of an Axiovert 200 M fluorescent microscope coupled to an AxioCamMR3 camera driven by the AxioVision 4.8 software (Carl Zeiss, North Ryde, Australia). The thickness of actin stress fibres was determined performing a line scan perpendicular to the fibres using Image J, while the size of paxillin-containing adhesion sites was measured using the AxioVision 4.8 software.
Colocalization analysis
Colocalization between β-actin and γ-actin channels was assessed by measuring the Pearson’s Correlation Coefficient and visually inspecting two-dimensional histograms (fluorograms). The Pearson’s coefficient measures the linear relationship between the pixel intensities of two channels. In the case of positive correlation this value can drop either due to decreasing colocalization, or due to differences in stoichiometry in structures. The fluorogram can distinguish these two scenarios, low colocalization shows by dispersion of points whilst varying stochiometries show multiple tight linear clusters. Measurements were made using the Coloc 2 plugin in ImageJ (imagej.nih.gov/ij). Image background was carefully subtracted from each channel and regions of interest were drawn around each cell to exclude extracellular pixels from the measurement. Measurements were performed on single cells to ensure variations in expression and staining did not contribute to multiple stoichiometries.
Adhesion assay
For the adhesion assay, cells were pre-labeled
Chemotaxis assay
The chemotaxis assay was performed as previously described [18]. Briefly, the underside of 8 μm transparent polyethylene terephthalate membrane inserts (BD Falcon) was pre-coated with 0.1% gelatin for 1 h. The cells were pre-labeled
Random motility assay
Random cell motility was assessed by time-lapse microscopy as previously described [18]. Briefly, cells were seeded on a 24-well gelatin-coated plate and allowed to adhere for 1 h. Photographs were then taken every 5 min for 6 h in at least 2 view fields per well using the 5X objective of the same microscope device used for immunofluorescence experiments. During this assay, cells were constantly maintained at 37°C and 5% CO2. Analysis was performed using the tracking module of the AxioVision 4.8 software. At least 25 cells per view field were tracked for 6 h; cells undergoing division or apoptosis were excluded from analyses. The persistent random-walk model was used to characterize cell motility [19]. For each individual cell, the mean square displacement < D2 > was calculated from the following formula:<D2>=∑i=1Mdi2\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$ <{D}^2>={\displaystyle \sum_{i=1}^M{d}_i^2} $$\end{document}
where di is the displacement of a cell from its initial position at time level ti. The persistence time (P) (i.e. average time interval between significant movements and direction changes) and random motility coefficient (μ) (i.e. the rate at which a cell population is able to migrate into and colonize a new area) were deduced from the < D2 > value and the cell velocity (S), using the following formulas:<D>=2S2P21/P−1+exp−1/Pμ=1/2S2P\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$ \begin{array}{c}\hfill <D>=2{\mathrm{S}}^2{\mathrm{P}}^2\left[\left(1/\mathrm{P}\right)-1+ \exp \left(-1/\mathrm{P}\right)\right]\hfill \\ {}\hfill \upmu =\left(1/2\right){\mathrm{S}}^2\mathrm{P}\hfill \end{array} $$\end{document}
Wound healing assay
An optimized wound healing assay was used as previously described [20], with slight modifications. Endothelial cells were grown to confluence in specific culture inserts (Ibidi, Martinsried, Germany). After 24 h, the culture inserts were removed, leaving a definite cell-free gap of approximately 400 μm, and the cells were washed with PBS before their incubation in culture medium. The colonization of the cell-free gap was analysed by time-lapse videomicroscopy using the 5X objective of the same microscope device used for immunofluorescence experiments. Photographs were taken every 10 minutes for 20 h and plates were kept at 37°C and 5% CO2 throughout the duration of the experiment. The migration rate was calculated digitally by quantification of the cell-free area at the different time points using the AxioVision 4.7 software.
In vitro Matrigel™ assay
Matrigel™ (BD Biosciences, North Ryde, Australia) assay was used to determine the effect of γ-actin knockdown on endothelial cell morphogenesis into capillary tubes, as previously described [18]. Briefly, 24-well plates were coated at 4°C with 270 μL of a Matrigel™ solution (1:1 dilution in culture medium), which was then allowed to solidify for 1 h at 37°C before cell seeding. Cells were allowed to undergo morphogenesis and form capillary-like structures and photographs were taken after 8 h using the 5X objective of the same microscope device used for immunofluorescence experiments. Angiogenesis was then quantitatively evaluated by measuring the total surface area of capillary tubes formed in at least 10 view fields per well using the AxioVision 4.7 software.
A non-enzymatic methodology was also established to analyse the potential changes in protein expression that occur during the morphological differentiation of endothelial cells into vascular networks. Briefly, 3.2 × 105 cells were seeded onto 6-well plates previously coated with Matrigel™ and harvested at different time points of the morphological differentiation process (i.e. 15 min, 1, 2, 4 and 8 h). Cells were first incubated with a Cell Recovery Solution (BD Biosciences) for 1 h at 4°C under agitation to allow complete dissolution of the Matrigel™, then pelleted, washed with cold PBS and finally lysed as described in the western blotting section.
Rho-associated kinase (ROCK) signalling inhibition
ROCK signalling was interrupted as previously described [8], through the use of two specific ROCK inhibitors, H-1152 (Merck Millipore, Kilsyth, Australia) and Y-27632 (Sigma-Aldrich). Stock solutions of both inhibitors were prepared in water and stored at 4°C. Inhibitors (1–10 μM) were added to siRNA-transfected cells at 48 h post-transfection, and remained in culture medium for a further 24 h and during wound-healing and angiogenesis assays.
Statistical analysis
All experiments were performed at least in triplicate. Statistical significance was determined using two-sided student’s t test in the GraphPad Prism 4 software (GraphPad Software, Inc).
Results
Spatial distribution of β- and γ-actin in vascular endothelial cells
Using specific monoclonal antibodies directed against β- and γ-actin [7], we investigated the cellular distribution of both actin isoforms in two models of vascular endothelial cells (Figure 1). These co-immunofluorescence experiments demonstrated extensive colocalization of the two actin isoforms with some level of spatial preference. In HMEC-1 and BMH29L endothelial cells, β- and γ-actin signals strongly overlapped but β-actin signal appeared relatively more enriched in radial stress fibres and membrane ruffling in relation to γ-actin, which was more uniformly spread across the entire microfilament meshwork. Quantification using the Pearson’s Correlation Coefficient method [21] confirmed the strong colocalization of β- and γ-actin (Additional file 1: Figure S1A). Furthermore, decreased correlation in some cells was mostly due to variations in stoichiometry in different subcellular structures rather than complete segregation of the two actin isoforms (Additional file 1: Figure S1B).
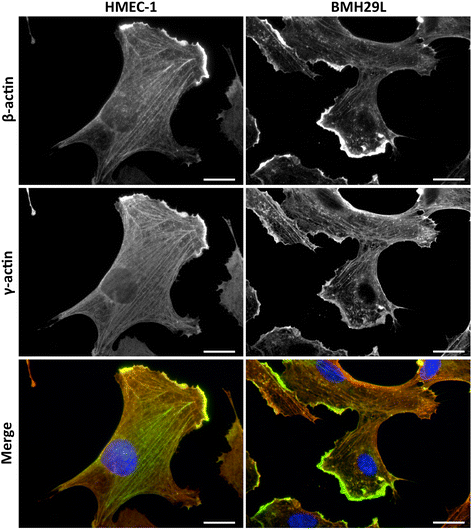
Figure 1
Figure 1 caption
Localization ofβ- andγ-actin in vascular endothelial cells. Representative photographs of HMEC-1 () and BMH29L () endothelial cells stained with β-actin () and γ-actin () antibodies. The merged photographs () show β-actin in green, γ-actin in red and DNA (DAPI) in blue. , 20 μm.
Knockdown of cytoplasmic γ-actin expression by RNAi
Significant knockdown of β-actin expression could not be achieved in endothelial cells without major cytotoxicity (data not shown). We therefore focused our attention on the functional analysis of γ-actin using RNA interference. As shown in Figure 2A, when HMEC-1 cells were transfected with γ-actin siRNA for 24 h, a 71 ± 12% reduction in γ-actin gene expression was observed by quantitative RT-PCR (p < 0.01). Consistently, a 50-60% knockdown of γ-actin expression was observed at the protein level after 72 h transfection in both HMEC-1 and BMH29L cells (Figure 2B and C; p < 0.001). Western blot analysis also showed that this level of γ-actin knockdown could be achieved without any significant compensatory changes in β-actin expression.
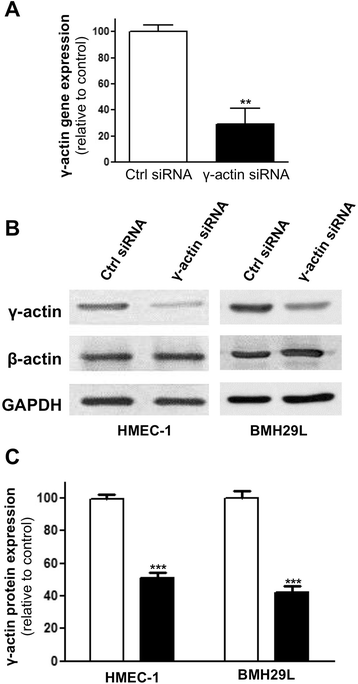
Figure 2
Figure 2 caption
γ-actin knockdown in vascular endothelial cells. (A) Histogram showing γ-actin relative gene expression following treatment with control () and γ-actin siRNA () for 24 h as assessed by quantitative RT-PCR. β was used as housekeeping gene. , means of at least three individual experiments; , SE. Statistics were calculated by comparing γ-actin relative expression in control and γ-actin siRNA-treated HMEC-1 cells; **, p < 0.01. (B) Representative immunoblots of HMEC-1 () and BMH29L () cell lysates following treatment with control and γ-actin siRNA for 72 h. Membranes were probed with anti-β-actin, anti-γ-actin and anti-GAPDH (loading control) antibodies. (C) Histogram showing the relative protein expression γ-actin as determined by densitometry after normalization with GAPDH, following treatment with control () and γ-actin siRNA () for 72 h. , means of at least four individual experiments; , SE. Statistics were calculated by comparing γ-actin expression level in control and γ-actin siRNA-treated cells; ***, p < 0.001.
Co-immunofluorescence staining was then used to analyse the effects of γ-actin knockdown on the localization of actin isoforms. In γ-actin siRNA-treated HMEC-1 cells, qualitative data revealed that the dense cortical actin meshwork was partially depleted in γ-actin and the remaining γ-actin was mostly found in stress fibres (Figure 3). In contrast, β-actin distribution was unaltered. Similar effects were observed in BMH29L cells (Additional file 2: Figure S2). Interestingly, quantitative analysis showed that γ-actin knockdown resulted in a slight increase in correlation of the β- and γ-actin signals, which was mostly due to decreased variation in stoichiometry (Additional file 1: Figure S1; Pearson coefficient of 0.75 ± 0.02 and 0.86 ± 0.01 in control and γ-actin siRNA-treated cells, respectively; p < 0.001).
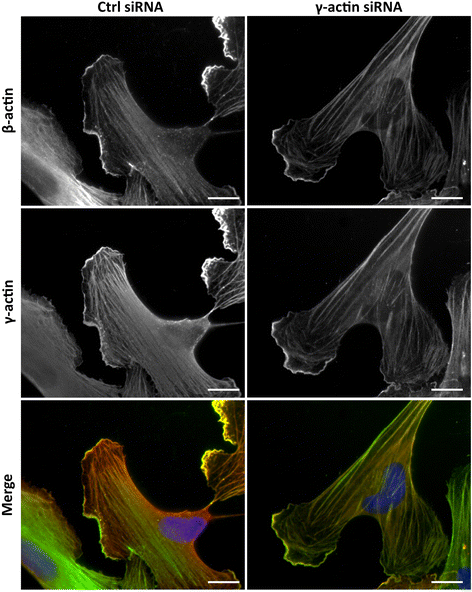
Figure 3
Figure 3 caption
Effect ofγ-actin knockdown on the localization ofβ- andγ-actin. Representative photographs of HMEC-1 endothelial cells treated for 72 h with control () or γ-actin siRNA () and stained with β-actin () and γ-actin () antibodies. The merged photographs () show β-actin in green, γ-actin in red and DNA (DAPI) in blue. , 20 μm.
Cytoplasmic γ-actin expression is essential for the morphological differentiation of endothelial cells into vascular networks
Matrigel™ assay was used to investigate the role of γ-actin in angiogenesis
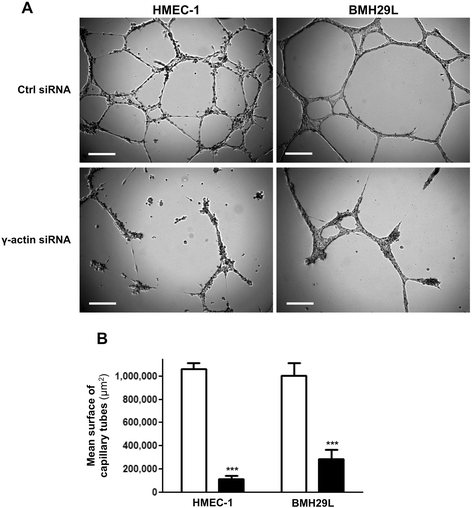
Figure 4
Figure 4 caption
Effect ofγ-actin knockdown on the formation vascular networks(A) Representative photographs of HMEC-1 () and BMH29L cells () incubated for 8 h on Matrigel™. Cells were treated either with control () or γ-actin siRNA () for 72 h. , 250 μm. (B) Histogram showing the surface occupied by vascular networks following treatment with control () and γ-actin siRNA () for 72 h. , means of at least four individual experiments; , SE. Statistics were calculated by comparing the mean surface occupied by vascular networks per view field (at least 10 view fields per condition) for control siRNA- and γ-actin siRNA-treated cells. ***, p < 0.001.
Vascular regression induced by γ-actin knockdown is associated with impaired VE-cadherin up-regulation
Endothelial cells were harvested non-enzymatically at different time points of the morphological differentiation process on Matrigel™ (Figure 5A) to investigate potential changes in expression of γ-actin, endothelial cell-to-cell contact protein VE-cadherin and major angiogenesis receptor VEGFR-2 by western blotting. While there was no significant change in γ-actin expression during the morphological differentiation of endothelial cells, VE-cadherin expression was gradually up-regulated (Figure 5B). Densitometry analysis revealed a 5-fold increase in VE-cadherin relative expression in control siRNA-treated cells between 15 min and 8 h incubation on Matrigel™ (Figure 5C; p < 0.001). Interestingly, γ-actin knockdown significantly impaired VE-cadherin up-regulation (Figure 5B and C). Although there was no significant difference in VE-cadherin expression between control siRNA- and γ-actin siRNA-treated cells at steady state when cells were grown on plastic (Additional file 5: Figure S4A), the levels of VE-cadherin expression were significantly lower in γ-actin siRNA-treated cells at all time points of the morphological differentiation process on Matrigel™ except at 4 h (Figure 5C). Additional western blot analysis further revealed that the expression of VEGFR-2 was also up-regulated during morphological differentiation, but this up-regulation was not affected by γ-actin knockdown (Additional file 5: Figure S4B). This suggests that the up-regulation of VE-cadherin expression, but not VEGFR-2, is dependent upon adequate γ-actin expression.
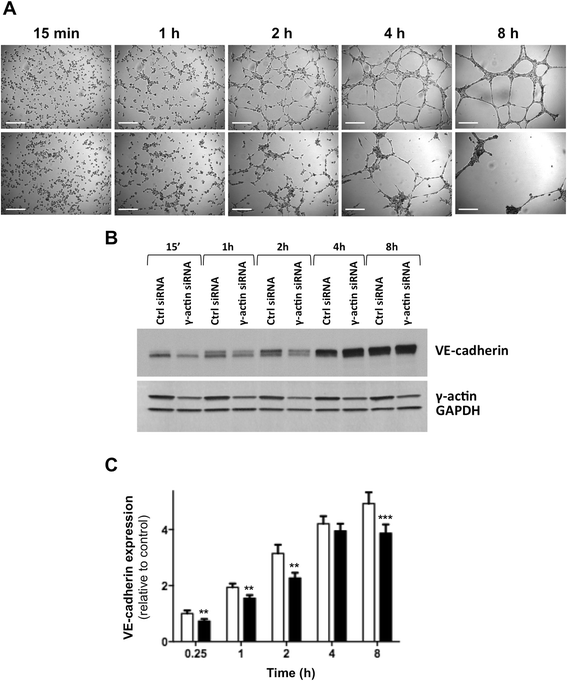
Figure 5
Figure 5 caption
Effect ofγ-actin knockdown on VE-cadherin expression during morphological differentiation of endothelial cells into vascular networks. (A) Representative photographs of HMEC-1 cells at various time points of the morphological differentiation process on Matrigel™, following treatment with control () and γ-actin siRNA () for 72 h. , 250 μm. (B) Representative immunoblots of HMEC-1 cell lysates obtained at different time points of the morphological differentiation process on Matrigel™, following treatment with control and γ-actin siRNA for 72 h. Membranes were probed with anti-VE-cadherin, anti-γ-actin and anti-GAPDH (loading control) antibodies. (C) Histogram showing the relative protein expression of VE-cadherin as determined by densitometry after normalization with GAPDH (loading control), following treatment with control () and γ-actin siRNA () for 72 h. , means of at least four individual experiments; , SE. Statistics were calculated by comparing VE-cadherin expression level in control and γ-actin siRNA-treated HMEC-1 cells; **, p < 0.01; ***, p < 0.001.
Cytoplasmic γ-actin plays a key role in endothelial cell motility and chemotaxis
To investigate the effects of γ-actin knockdown on the pro-angiogenic functions of endothelial cells, a panel of cell biology assays was used. First, we found that knocking down γ-actin expression did not significantly affect the adhesion of endothelial cells to various substrates, including major ECM proteins fibronectin, laminin and collagen I (Figure 6A). In contrast, knocking down γ-actin expression significantly impaired the chemotactic response of endothelial cells to various chemo-attractants. As shown in Figure 6B, addition of FCS, VEGF, FGFβ or ECGF in the bottom well of the Boyden chamber resulted in a significant increase in migration of control siRNA-treated HMEC-1 cells, as compared to the negative control (absence of FCS; p < 0.05). This increase in cell migration was significantly inhibited when HMEC-1 cells were transfected with γ-actin siRNA (p < 0.05). Interestingly, FGFβ-induced migration was completely suppressed by γ-actin knockdown, while migration induced by other chemo-attractants was only partially reduced.
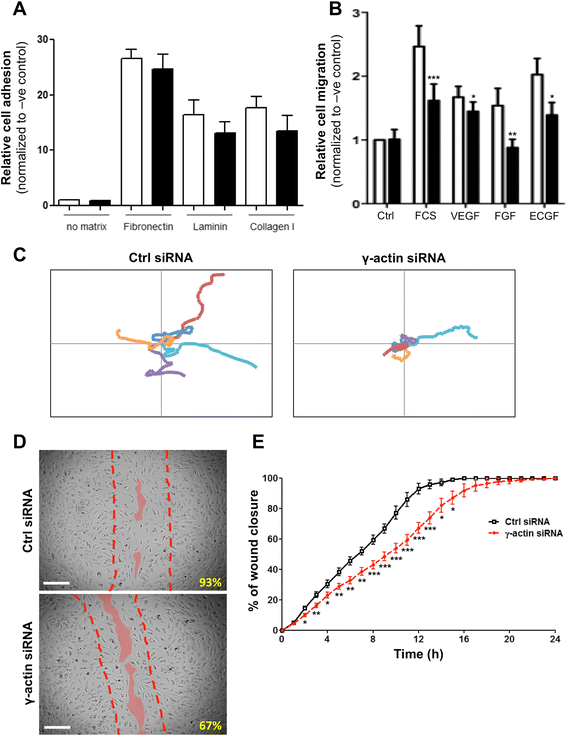
Figure 6
Figure 6 caption
Effect ofγ-actin knockdown on cell adhesion, migration and motility. (A) Histogram showing the relative adhesion of endothelial cells following treatment with control () and γ-actin siRNA () for 72 h. Fluorescently labeled HMEC-1 cells were allowed to adhere to various substrates for 1 h. , means of at least three individual experiments; , SE. (B) Histogram showing the relative migration of endothelial cells towards FCS, VEGF, FGF and ECGF following treatment with control () and γ-actin siRNA () for 72 h. Fluorescently labeled HMEC-1 cells were allowed to migrate through 8 μm pore PET membrane and towards a chemo-attractant for 6 h. , means of at least four individual experiments; , SE. Statistics were calculated by comparing the fluorescence measured at 492/517 (Abs/Em) with control and γ-actin siRNA-treated cells; *, p < 0.05; **, p < 0.001. (C) Representative trajectories of 5 individual HMEC-1 cells, recorded by time-lapse videomicroscopy over 6 h, following treatment with control () and γ-actin siRNA () for 72 h. , −130 μm to +130 μm for both x and y axes. (D) Representative photographs of control () and γ-actin siRNA-treated () BMH29L cells in wound healing experiments, taken 12 h after start of experiment. Broken lines show the position of the initial cell-free gap (at time 0) and solid lines highlight the position of the migration edge after 12 h. , % of wound closure. (E) Graph showing the percentage of wound recovery as a function of time for control () and γ-actin siRNA-treated () BMH29L cells. , means of at least four individual experiments; , SE. Statistics were calculated by comparing control and γ-actin siRNA-treated cells at specific time points; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
The effect of γ-actin knockdown on the random motility of endothelial cells was then assessed by time-lapse videomicroscopy experiments. Figure 6C shows the representative trajectories of 5 cells transfected with either control (
Table 1
Effects of γ-actin knockdown on random motility parameters
| Parameters | Control siRNA | γ-actin siRNA | % change |
|---|---|---|---|
| Average cell velocity (μm/min) | 0.53 ± 0.03 | 0.37 ± 0.03 | −30 ± 5 %** |
| Persistence time (min) | 6.3 ± 0.3 | 8.6 ± 0.6 | +36 ± 7 %* |
| Random motility coefficient (μm2/min) | 0.90 ± 0.05 | 0.55 ± 0.07 | −38 ± 8 %** |
In order to further investigate the effects of γ-actin knockdown on endothelial cell migration, wound healing experiments were performed using time-lapse videomicroscopy. Figure 6D shows representative photographs taken from control (
Cytoplasmic γ-actin regulates endothelial cell motility through ROCK signalling pathway
In order to gain more insights into the role of γ-actin in endothelial cell motility, we investigated potential changes in the organisation of microtubules, actin stress fibres and focal adhesions as a result of γ-actin knockdown. Tubulin staining of endothelial cells showed no significant change in the general organisation of the microtubule network as a result of γ-actin knockdown (Additional file 6: Figure S5). In contrast, co-immunofluorescence experiments with phalloidin and anti-paxillin antibody demonstrated that transfection of endothelial cells with γ-actin siRNA led to an accumulation of thick actin stress fibres and large paxillin-containing focal adhesions (Figure 7A). Quantitative analysis revealed that γ-actin knockdown induced a 38% increase in the average thickness of actin stress fibres, which was 0.75 ± 0.15 μm and 1.04 ± 0.17 μm in control and γ-actin siRNA-treated cells, respectively (Figure 7B; p < 0.001). This demonstrates that γ-actin knockdown results in a change in filamentous actin (F-actin) structures as a result of changing the β- to γ-actin ratio. Control siRNA-treated cells displayed uniform paxillin staining with small focal adhesions located throughout the cytoplasm and in the lamellipodia. In sharp contrast, γ-actin siRNA-treated cells showed larger focal adhesions dispersed throughout the cell periphery. Quantitative analysis showed that γ-actin knockdown did not alter the number of paxillin-containing adhesions but significantly increased their average size by 76%, from 0.96 ± 0.08 μm2 to 1.67 ± 0.11 μm2 (Figure 7C; p < 0.001). This accumulation of thick actin stress fibres and large focal adhesions could result from sustained activation of ROCK signalling [8]. Western blot analysis revealed that γ-actin knockdown was associated with a 58% increase in phosphorylation of myosin light chain 2 (Figure 7D-E), thus confirming activation of the ROCK pathway.
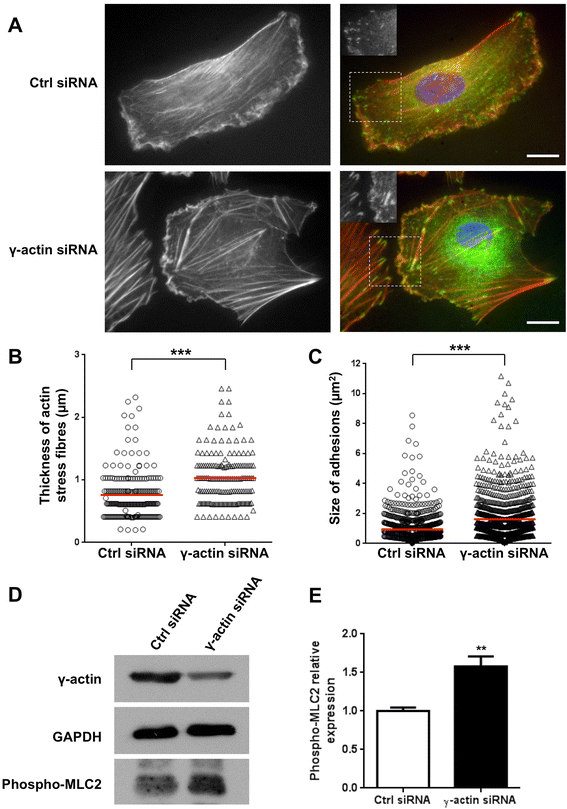
Figure 7
Figure 7 caption
Effect ofγ-actin knockdown on ROCK signalling. (A) Representative photographs of HMEC-1 endothelial cells treated for 72 h with control () or γ-actin siRNA () and co-stained with phalloidin () and anti-paxillin antibody. The merged photographs () show phalloidin in red, paxillin in green and DNA (DAPI) in blue. shows a magnified view of paxillin staining in the lamellipodial region. , 20 μm. (B-C) Scatter dot plots showing the thickness of actin stress fibres and the size of paxillin-containing adhesions in HMEC-1 endothelial cells treated for 72 h with control (o) or γ-actin siRNA (∆). , means of at least 15 individual cells. (D) Representative immunoblots of HMEC-1 cell lysates following treatment with control and γ-actin siRNA for 72 h. Membranes were probed with anti-γ-actin, anti-GAPDH (loading control) and anti-phospho-myosin light chain 2 antibodies. (E) Histogram showing the relative levels of phosphorylated myosin light chain 2 in HMEC-1 cells following treatment with control () and γ-actin siRNA () for 72 h. , means of at least four individual experiments; , SE. Statistics were calculated by comparing control siRNA- versus γ-actin siRNA-transfected cells. **, p < 0.01; ***, p < 0.001.
Cytoplasmic γ-actin regulates angiogenesis through ROCK-dependent and ROCK-independent mechanisms
Pharmacological inhibition of ROCK signalling showed that the impairment of motility induced by γ-actin knockdown was entirely dependent upon ROCK activation (Figure 8A). Indeed, incubation with H-1152 and Y-27632 inhibitors completely restored the motility of γ-actin siRNA-treated HMEC-1 cells. Similar results were obtained with BMH29L cells (Additional file 7: Figure S6). In contrast with the complete rescue of the γ-actin knockdown motility phenotype by ROCK inhibitors, the capacity of γ-actin siRNA-treated cells to form and maintain vascular networks on Matrigel™ was only partially restored by ROCK inhibition (Figure 8B-D). Incubation of control siRNA-treated HMEC-1 cells with H-1152 and Y-27362 increased vascular sprouting and therefore led to the formation of more but significantly smaller capillary-like tubes, resulting in a marginal decrease in total vascular surface (4-6% decrease as compared to control untreated cells). The angiogenic potential of γ-actin siRNA-treated cells was improved when ROCK signalling was inhibited but vascular regression was not entirely prevented (Figure 8B). In γ-actin knockdown HMEC-1 cells, angiogenesis inhibition was thus reduced from 75 ± 6% in untreated cells to 38 ± 8% and 26 ± 8% in cells incubated with H-1152 and Y-27632, respectively (Figure 8C; p < 0.001). Quantification of the number of vascular networks formed confirmed that incubation with H-1152 and Y-27362 increased vascular sprouting (Figure 8D; p < 0.001) but γ-actin knockdown still resulted in significantly less networks formed in all conditions. Partial rescue of the angiogenesis suppression induced by γ-actin knockdown was also observed in BMH29L cells as a result of ROCK inhibition (Additional file 8: Figure S7). Collectively, the results of our study show that γ-actin plays a key role in the morphological differentiation of endothelial cells into vascular networks, through both ROCK-dependent and ROCK-independent mechanisms.
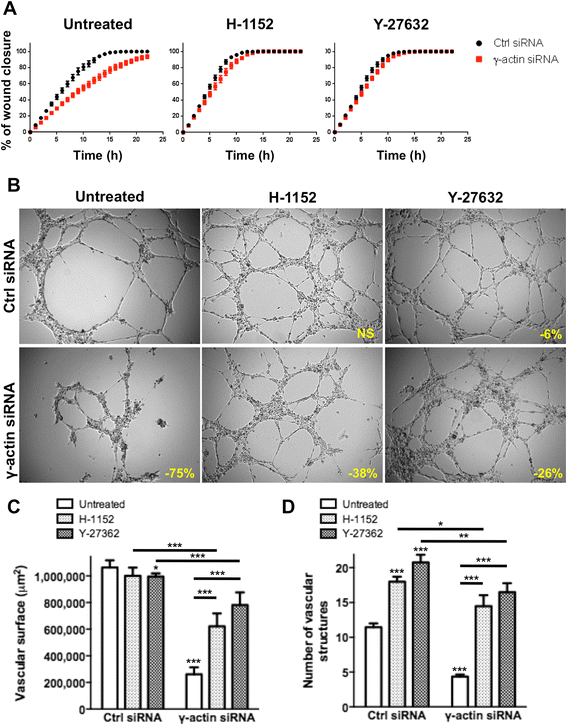
Figure 8
Figure 8 caption
Effect of ROCK signalling inhibition onγ-actin knockdown-induced vascular regression. (A) Graph showing the percentage of wound recovery as a function of time for control () and γ-actin knockdown () HMEC-1 cells, either untreated () or treated with ROCK inhibitors, H-1152 () or Y-27632 () at 1 and 10 μM, respectively. , means of at least four individual experiments; , SE. (B) Representative photographs of HMEC-1 cells incubated for 8 h on Matrigel. Cells were transfected with either control () or γ-actin siRNA () for 72 h and either untreated () or treated with ROCK inhibitors, H-1152 () or Y-27632 () at 1 and 10 μM, respectively. , % of angiogenesis inhibition as compared to untreated control cells. , 250 μm. (C-D) Histograms showing the surface occupied by and the number of vascular networks formed by control and γ-actin siRNA-transfected HMEC-1 cells following treatment with ROCK inhibitors. , means of at least four individual experiments; , SE. Statistics were calculated by comparing the mean surface occupied by vascular networks and the mean number of vascular networks per view field (at least 10 view fields per condition) for control siRNA- versus γ-actin siRNA-transfected cells unless indicated otherwise. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
Actin-binding and regulatory proteins have been proposed to represent attractive targets for the development of innovative anti-angiogenic therapies [22]. However, the role of the actin cytoskeleton in angiogenesis has not been completely elucidated. Basic cell biology studies are crucially needed to map the specific role played by each component of the actin cytoskeleton in order to find new ways to target angiogenesis in pathologies such as cancer and rheumatoid arthritis. Here, we investigated the cellular distribution and localization of the two cytoplasmic isoforms of actin, β and γ, in vascular endothelial cells and performed the first functional analysis of γ-actin in these cells, revealing a key role for this protein in angiogenesis.
Co-immunofluorescence staining using newly developed antibodies directed against β- and γ-actin revealed a strong colocalization of the two cytoplasmic actin isoforms in vascular endothelial cells, albeit some level of spatial preference. Our findings are in accordance with the preferential localization of β-actin in radial stress fibres and membrane ruffling and γ-actin in the cortical microfilament meshwork previously reported in epithelial cells, fibroblasts [7] and more recently in neuroblastoma cells [8]. The subcellular localization of β- and γ-actin expression appears to be mediated by localization of their respective mRNA [5]. Furthermore, studies have shown that β- and γ-actin differentially interact with accessory proteins such as tropomyosins [23] and non-sarcomeric myosins [24]. These differential interactions may govern the specific functions of β- and γ-actin, and therefore contribute to the spatial and temporal regulation of cytoskeletal dynamics.
Significant knockdown of β-actin could not be achieved in vascular endothelial cells without inducing major cytotoxicity, suggesting that these cells are particularly sensitive to changes in β-actin expression. In contrast, γ-actin expression could be successfully knocked down by 50-60% in endothelial cells, indicating an isoform-specific function for actin in endothelial cells. This difference in response to β- and γ-actin knockdown is consistent with recent studies showing that β-actin knockout mice are not viable [9] unlike γ-actin knockout mice, which despite suffering increased mortality and showing progressive hearing loss during adulthood, are viable [10].
Interestingly, γ-actin knockdown almost completely suppressed the capacity of endothelial cells to maintain capillary-like structures on Matrigel™. Timelapse videomicroscopy analysis revealed that γ-actin was dispensable in the early steps of the morphological differentiation process (i.e. vascular sprouting) but required to sustain newly formed vascular networks. Previous studies have shown that the major endothelial cell adhesion molecule, VE-cadherin, plays a crucial role in angiogenesis – reviewed in [25-27]. The development of a VE-cadherin knockout (
To better understand how γ-actin knockdown impaired the angiogenic process, we investigated its impact on endothelial cell adhesion, chemotaxis and motility. While partially depleting γ-actin expression did not alter cell adhesion, it significantly reduced the capacity of endothelial cells to respond to chemotactic stimuli, randomly explore their microenvironment and colonize a new area. This result is in line with other studies demonstrating the crucial role played by the actin cytoskeleton in cell locomotion in non-endothelial cells – reviewed by Le Clainche and Carlier [34]. It is also consistent with the results recently reported for γ-actin depleted epithelial cells [7], arguing in favour of a crucial role of γ-actin in the control of directional motility in diverse cell types.
We previously reported that γ-actin regulates neuroblastoma cell migration by modulating ROCK signalling pathway [8]. Here, we extended this finding to vascular endothelial cells. Indeed, γ-actin knockdown was associated with accumulation of thicker actin stress fibres, larger focal adhesions and increased phosphorylation of myosin light chain 2 in endothelial cells, thus strongly suggesting activation of the ROCK signalling pathway. Furthermore, incubation with pharmacological inhibitors of ROCK signalling completely restored the motility of γ-actin siRNA-treated cells, demonstrating that the γ-actin knockdown motility phenotype is dependent upon ROCK activation. In contrast, ROCK inhibitors only partially rescued the angiogenic potential of γ-actin knockdown cells, suggesting that γ-actin regulates angiogenesis through both ROCK-dependent and ROCK-independent mechanisms.
Interestingly, VE-cadherin engagement has been shown to activate numerous biochemical pathways that can affect cell shape and adhesion, including the ROCK signalling pathway [35-37]. Furthermore, a direct link was recently reported between actin stress fibres and the protein complex forming adherens junctions, VE-cadherin/β-catenin/α-catenin [38,39]. Here we provide evidence of the reverse relationship since decreasing the expression of γ-actin partially blocked the required up-regulation of VE-cadherin expression during morphological differentiation of endothelial cells. Additional experiments are currently underway to fully decipher the mechanisms involved in the crosstalk between γ-actin, ROCK signalling and VE-cadherin, which appears to play a critical role in the maintenance of newly formed vascular networks.
Conclusions
Collectively our results demonstrate for the first time that γ-actin plays a crucial role in angiogenesis through the regulation of endothelial cell motility and neovessel maintenance,
Acknowledgments
The authors would like to thank Dr Christine Chaponnier (University of Geneva, Switzerland) for generously providing the β- and γ-actin monoclonal antibodies and Dr Karen MacKenzie (Children’s Cancer Institute Australia) for providing the BMH29L endothelial cell line. This work was supported by the Children’s Cancer Institute Australia for Medical Research, which is affiliated with the University of New South Wales and Sydney Children’s Hospital, and by grants from the Cancer Institute New South Wales (EP) and Cancer Council New South Wales (MK). EP was supported by a Cancer Institute New South Wales “Early Career Development” Fellowship. MK is supported by a NHMRC Senior Research Fellowship (APP1058299) and an Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology.
Additional files
Quantification of β- and γ-actin colocalization. (A) Scatter dot plot showing the Pearson’s coefficient of individual HMEC-1 cells treated for 72 h with control (o) or γ-actin siRNA (∆). Bars, mean of three individual experiments with SE. Statistics were calculated by comparing control siRNA- versus γ-actin siRNA-transfected cells. ***, p < 0.001. (B) Representative 2-Dimensional histograms (fluorograms) showing a single linear relationship in high correlating samples (left) and multiple linear relationships in lower correlating cells (right). This shows that the lower correlation of β-actin and γ-actin signals in some cells was mostly due to variations in stoichiometry rather complete segregation of the two isoforms.
Effect of γ-actin knockdown on the actin cytoskeleton. Representative photographs of BMH29L endothelial cells treated for 72 h with control (left) or γ-actin siRNA (right) and stained with β-actin (top) and γ-actin (middle) antibodies. The merged photographs (bottom) show β-actin in green, γ-actin in red and DNA (DAPI) in blue. Scale bar, 20 μm.
Effect of γ-actin knockdown on the formation vascular networks on Matrigel™ using a different siRNA sequence. (A) Representative immunoblots of HMEC-1 cell lysates following treatment with control and γ-actin siRNA2 for 72 h. Membranes were probed with anti-β-actin, anti-γ-actin and anti-GAPDH (loading control) antibodies. (B) Histogram showing the relative protein expression of γ-actin as determined by densitometry after normalization with GAPDH (loading control), following treatment with control (white) and γ-actin siRNA2 (black) for 72 h. Columns, means of three individual experiments; bars, SE. Statistics were calculated by comparing γ-actin expression level in control and γ-actin siRNA2-treated HMEC-1 cells; ***, p < 0.001. (C) Representative photographs of HMEC-1 cells incubated for 8 h on Matrigel™ and following treatment with either control (left) or γ-actin siRNA (bottom) for 72 h. Scale bar, 250 μm. (D) Histogram showing the surface occupied by vascular networks following treatment with control (white) and γ-actin siRNA (black) for 72 h. Columns, means of three individual experiments; bars, SE. Statistics were calculated by comparing the mean surface occupied by vascular networks per view field (at least 10 view fields per condition) for control and γ-actin siRNA-treated HMEC-1 cells. ***, p < 0.001.
Supplementary videos 1–2. Representative time-lapse videomicroscopy experiments of control siRNA- and γ-actin siRNA-treated endothelial cells on Matrigel™. BMH29L cells were seeded on Matrigel™ 72h after transfection with either control (Video 1) or γ-actin siRNA (Video 2). Photographs were taken every 5 min for 12 h, using the 5X objective of an Axiovert 200M fluorescent microscope coupled to an AxioCamMR3 camera driven by the AxioVision 4.7 software.
Effect of γ-actin knockdown on VE-cadherin and VEGFR-2 expression at steady state and during the angiogenic process. (A) Representative immunoblots of HMEC-1 (left) and BMH29L (right) cell lysates following treatment with control and γ-actin siRNA for 72 h. Membranes were probed with anti-VEGFR-2, anti-VE-cadherin, anti-γ-actin and anti-α-tubulin (loading control) antibodies. (B) Representative immunoblots of HMEC-1 cell lysates obtained at different time points of the morphological differentiation process on Matrigel™, following treatment with either control (left) or γ-actin siRNA (right) for 72 h. Membranes were probed with anti-VEGFR-2, anti-γ-actin and anti-GAPDH (loading control) antibodies.
Effect of γ-actin knockdown on the microtubule network. Representative photographs of HMEC-1 cells treated for 72 h with control (left) and γ-actin siRNA (right) and stained with anti-tubulin antibody. Scale bar, 20 μm.
Effect of ROCK signalling inhibition on γ-actin knockdown-induced motility inhibition. Representative photographs of wound healing experiments with control and γ-actin siRNA-transfected BMH29L cells. Endothelial cells were either untreated (left) or pre-treated with H-1152 (middle) or Y-27632 (right) at 10 μM for 24 hours and during the course of the experiment. Broken lines show the position of the initial cell-free gap (at time 0) and solid lines highlight the position of the migration edge after 8 h. Inset, % of wound closure.
Effect of ROCK signalling inhibition on γ-actin knockdown-induced vascular regression. (A) Representative photographs of BMH29L cells incubated for 8 h on Matrigel™. Cells were transfected with either control (top) or γ-actin siRNA (bottom) for 72 h and either untreated (left) or treated with 10 μM ROCK inhibitors, H-1152 (middle) or Y-27632 (right). Inset, % of angiogenesis inhibition as compared to untreated control cells. Scale bar, 250 μm. (B) Histogram showing the surface occupied by vascular networks following treatment with control (white) and γ-actin siRNA (black) for 72 h. Columns, means of at least four individual experiments; bars, SE. Statistics were calculated by comparing the mean surface occupied by vascular networks per view field (at least 10 view fields per condition) for control siRNA- vs γ-actin siRNA-transfected cells unless indicated otherwise. *, p<0.05; ***, p < 0.001.
References
- Angiogenesis in life, disease and medicine. Nature. 2005;438:932-936.
- Angiogenesis as a therapeutic target. Nature. 2005;438:967-974.
- Targeting microtubules to inhibit angiogenesis and disrupt tumour vasculature: implications for cancer treatment. Curr Cancer Drug Targets. 2007;7:566-581.
- Actin isoform expression patterns during mammalian development and in pathology: insights from mouse models. Cell Motil Cytoskeleton. 2009;66:798-815.
- Beta and gamma actin mRNAs are differentially located within myoblasts. J Cell Biol. 1993;122:825-832.
- Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci. 1998;18:251-265.
- Beta and gamma-cytoplasmic actins display distinct distribution and functional diversity. J Cell Sci. 2009;122:2980-2988.
- gamma-Actin regulates cell migration and modulates the ROCK signaling pathway. FASEB J. 2011;25:4423-4233.
- β-Actin specifically controls cell growth, migration, and the G-actin pool. Mol Biol Cell. 2011;22:4047-4058.
- Gamma-actin is required for cytoskeletal maintenance but not development. Proc Natl Acad Sci U S A. 2009;106:9703-9708.
- HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683-690.
- Multiple stages of malignant transformation of human endothelial cells modelled by co-expression of telomerase reverse transcriptase, SV40 T antigen and oncogenic N-ras. Oncogene. 2002;21:4200-4211.
- Alterations in gamma-actin and tubulin-targeted drug resistance in childhood leukemia. J Natl Cancer Inst. 2006;98:1363-1374.
- Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci. 2001;114:4557-4565.
- Concentration- and schedule-dependent effects of chemotherapy on the angiogenic potential and drug sensitivity of vascular endothelial cells. Angiogenesis. 2012;16:373-386.
- Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal Biochem. 1999;270:41-49.
- Tissue-specific tropomyosin isoform composition. J Histochem Cytochem. 2005;53:557-570.
- ENMD-1198, a new analogue of 2-methoxyestradiol, displays both antiangiogenic and vascular-disrupting properties. Mol Cancer Ther. 2010;9:1408-1418.
- Endothelial cell migration on surfaces modified with immobilized adhesive peptides. Biomaterials. 2000;21:1725-1733.
- Effects of bortezomib on human neuroblastoma cells in vitro and in a metastatic xenograft model. Anticancer Res. 2009;29:1219-1225.
- Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry A. 2010;77:733-742.
- Novel approaches in anti-angiogenic treatment targeting endothelial F-actin: a new anti-angiogenic strategy?. Curr Opin Mol Ther. 2008;10:579-590.
- Differential regulation of tropomyosin isoform organization and gene expression in response to altered actin gene expression. J Cell Biol. 1993;121:811-821.
- Distinct functional interactions between actin isoforms and nonsarcomeric myosins. PLoS One. 2013;8:e70636-.
- Endothelial cadherins and tumor angiogenesis. Exp Cell Res. 2006;312:659-667.
- Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb Res. 2007;120:S1-S6.
- VE-cadherin: the major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol. 2008;28:223-232.
- Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147-157.
- VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood. 2005;105:2771-2776.
- The human VE-cadherin promoter is subjected to organ-specific regulation and is activated in tumour angiogenesis. Oncogene. 2005;24:2992-3001.
- VE-cadherin-mediated cell-cell interaction suppresses sprouting via signalling to MLC2 phosphorylation. Curr Biol. 2009;19:668-674.
- The myosin motor Myo1c is required for VEGFR2 delivery to the cell surface and for angiogenic signalling. Am J Physiol Heart Circ Physiol. 2013;304:H687-H696.
- Inhibition of Rac1 GTPase downregulates vascular endothelial growth factor receptor-2 expression by suppressing Sp1-dependent DNA binding in human endothelial cells. Exp Dermatol. 2009;18:863-869.
- Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 2008;88:489-513.
- VE-cadherin regulates endothelial actin activating Rac and increasing membrane association of Tiam. Mol Biol Cell. 2002;13:1175-1189.
- VE-cadherin-induced Cdc42 signaling regulates formation of membrane protrusions in endothelial cells. J Biol Chem. 2003;278:16230-16236.
- Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell. 2004;15:2943-2953.
- Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol. 2010;8:11-.
- Vascular endothelial-cadherin stabilizes at cell-cell junctions by anchoring to circumferential actin bundles through alpha- and beta-catenins in cyclic AMP-Epac-Rap1 signal-activated endothelial cells. Mol Biol Cell. 2010;21:584-596.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.