An update on angiogenesis modulators with a potential therapeutic role
Vascular Cell. 2018;
Received: 12 August 2018 | Accepted: 2 December 2018 | Published: 22 December 2018
Vascular Cell ISSN: 2045-824X
Abstract
Angiogenesis or formation of new blood vessels as a consequence of "sprouting" in a pre-existent vascular network represents a phenomenon of paramount importance in physiology and pathology. Although the molecular pathways and factors which modulate angiogenesis are similar regardless the clinical context, there are numerous aspects related to this process which are incompletely understood. At the present time, significant research focuses in finding the best method to control the pro-angiogenic factors as well as anti-angiogenesis since therapy of various medical conditions requires either stimulation or inhibition of formation of new blood vessels. However, angiogenesis is different in various vascular systems and organs. In this review, we will present the most promising factors that interfere with angiogenesis suggesting new or improved therapeutic methods in various pathological settings.
Keywords
SDF1 HIF1 MMPTie-2 Notch WNT CCNA1 PTEN AGEs/RAGEs HMGB1Introduction
Blood vessels are formed during gastrulation stage by fibroblast growth factor (FGF) which acts upon the mesoderm to create accumulations of hematopoietic cells and angioblasts called “blood islands of the yolk sac”. The angioblasts are in fact primitive endothelial cells. However, vascular endothelial growth factor (VEGF) promotes further differentiation of primitive vascular tubules from these angioblasts [1]. At this stage, the vascular structures are immature and friable but additional fibroblast growth factor (FGF)-dependent synthesis of vascular endothelial growth factor (VEGF) and its receptor would subsequently drive the formation of mature blood vessels [2]. This step is characterized by the acquisition of a mural cell coat which offers functional stability for the immature embryonic vessels and concludes the process of angiogenesis [3, 4]. In several pathological settings, including in those associated with ischemia, angiogenesis requires the mobilization of bone marrow-precursor cells and their differentiation into circulating endothelial precursor cells. In ischemic lesions, it is described a direct attachment of these endothelial precursor cells to form the endothelium of capillaries located in the ischemic areas. Subsequently, this leads to the revascularization of affected tissues [5]. From a mechanistic perspective, regardless the clinical setting angiogenesis represents a multi-step process which begins with dilatation of pre-existing vessels and a vascular endothelial growth factor (VEGF)-dependent activation of endothelial cells. The next step consists of synchronous metalloproteinase-dependent capillary basement membrane disassembly and breakage of inter-cellular endothelial bunches mediated by a plasminogen activator. This results in a significant increased vascular permeability. In parallel, additional degradation of peri-capillary matrix components, modulated by the same pro-angiogenic factors acts in concert with migration and proliferation of endothelial cells to create endothelial sprouts. Further organization of these sprouts leads to the formation of a network of endothelial cords. Depending on the interaction between various pro-angiogenic factors the ultimate result will be represented by vascular remodelling of this network with the formation of vascular tubular structures. Maturation of these young vessels involves recruitment and proliferation of pericytes and formation of a new basement membrane and some smooth muscle fibres layer. Meantime, other factors such as transforming growth factor beta (TGF-b) can initiate differentiation of microvascular endothelial cells and pericytes into matrix-depositing myofibroblasts. Finally, after fusion of the newly formed vessels, they become functional and blood-flow is initiated through the vascular network [6, 7, 8] (Figure 1).
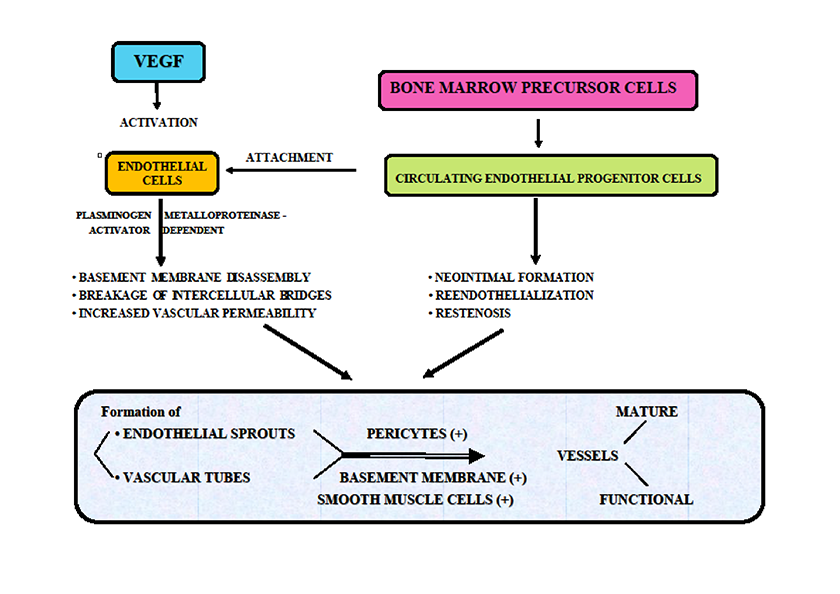
Figure 1
Figure 1 caption
Angiogenesis: VEGF and main factors
However, despite all of these, the process of angiogenesis is not completely understood. Meantime, current therapy of cancer or diabetes requires new improved anti-angiogenic pharmacological agents while in other pathological entities such as stroke an efficient manipulation of angiogenesis is yet to be implemented in a clinical setting. In this current article, we will present the most promising factors which interfere with angiogenesis in various clinical settings.
a. Stromal Cell-derived Factor 1 (SDF1)
Research studies investigating the angiogenic modulator role of Stromal Cell-derived Factor 1 (SDF1) have been conducted for quite some time but unfortunately, its complex actions are still incompletely understood. Initially, Mirshahi et al. (2000) have shown in an experimental study that Stromal Cell-derived Factor 1 (SDF1) acts upon C-X-C chemokine receptor 4 (CXCR4) and C-X-C Motif Chemokine Ligand 12 (CXCL12) and has a significant pro-angiogenic effect in various experimental models [9]. The authors have shown that this factor is pro-angiogenic in vitro in a tridimensional fibrin gel promoting VEFG synthesis by endothelial cells. Also, SDF-1 has a pro-angiogenic effect in vivo in a model of rabbit corneal pocket [9]. In patients with glioblastoma multiforme, SDF-1/CXCL12 expression increases with increasing grade of the tumour and is better identified in more vascularized areas of the tumour [10]. It seems that the pro-angiogenic action of SDF-1 is enhanced by heme-oxygenase 1 (HO-1) which is locally manufactured by endothelial cells via protein kinase C zeta-dependent activation. Animal studies revealed that HO-1 deficient animals present with abnormal healing of the retinal blood vessels after SDF-1 administration compared with those subjects with HO-1 normal profile suggesting that HO-1 may act as a co-factor in this pathway [11].
Remarkably, SDF-1 is chemotactic and chemoattractant for a selected group of bone marrow cells bearing the C-X-C chemokine receptor 4 (CXCR4) which might be involved in tumour development as well as revascularization after ischemia. However, the actual mechanism of angiogenesis mediated by the interaction between SDF-1 and CXCR4 positive bone marrow cells is yet poorly understood. In experimental models of ischemia, apart from the mobilization of CXCR4 positive bone marrow cells, bone marrow cells that express VEGF receptors are also activated by SDF-1 [12]. Experiments conducted in patients with critical leg ischemia have shown that SDF-1 presents with two spliced forms alpha and beta. However, it seems that the beta variant has a more potent pro-angiogenic effect but both forms activate PI3K/Akt and p44/42 kinase [13].
In another pathological setting, SDF-1 partially counteracts the anti-angiogenic action of LDL. However, LDL decreases CXCR4 receptors on the surface of endothelial cells but SDF-1 administered in parallel with VEGF in Matrigel-based tube formation assays-HUVECs, induces a full restoration of angiogenesis as well as a sustained anti-oxidative effect of endothelial cells suggesting that other molecular pathways might be involved [14].
Recent studies have also shown that in vitro SDF-1 promotes angiogenesis via activation of CXCR7 in a dose-dependent manner. Moreover, this mechanism stimulates the formation of tubules in HUVECs and in vivo systems. Remarkable, SDF-1/CXCL12 complex has a unidirectional chemoattractant action in HUVECs. It seems that CXCR7 action is facilitated by the PI3K/Akt phosphorylation pathway suggesting that SDF-1/CXCR7 represents a complex receptor for Akt-induced angiogenesis [15].
In vitro studies conducted in endothelial cells by Ziegler and Hughes (2014) indicate that SDF-1/CXC12 complex activates mTOR while inhibition of CXCR4 and mTOR inhibits in vitro 3D angiogenesis [16]. Therefore, SDF-1 could be a co-factor which acts in concert with other mediators upon the same types of receptors. Studies conducted in rats with experimental carotid artery and intracranial aneurysms have indicated that SDF-1 administration is associated not only with a pro-angiogenic effect but also with a deleterious inflammatory reaction in the walls of both types of aneurysms which may lead to subsequent rupture [17]. However, other groups have reported that SDF-1 alpha overexpression in bone marrow-derived stromal cells promotes healing in osteonecrosis of the femoral head as well as angiogenesis [18].
b. Hypoxia-inducible factors (HIF)
Hypoxia is responsible for the onset of angiogenesis, through releasing of the pro-angiogenic hypoxia-inducible transcription factors HIF1 and HIF2. Particularly, HIF1 induces trans-activation of gene-promoting angiogenesis and post-transcriptional stabilization of VEGF mRNA, followed by up-regulation of VEGF and other pro-angiogenic substances [20, 21, 22] (Figure 2).
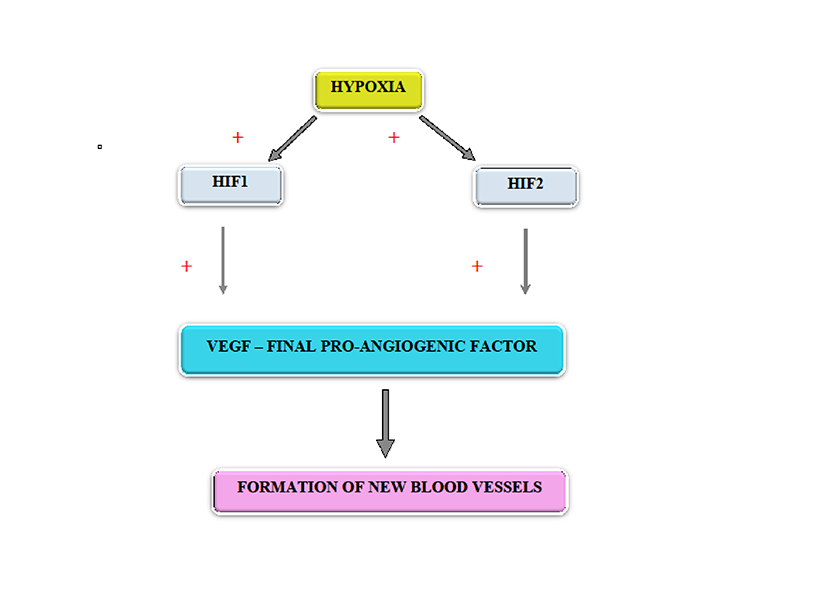
Figure 2
Figure 2 caption
Modulation of angiogenesis by hypoxia
At the level of the central nervous system, it seems that the most important mechanism modulating the extent of the vascular network is represented by an interplay between angiolysis and angiogenesis induced by HIFs acting in concerts with VEGF as well as the Ang2/PGE2/COX2 complex [19]. However, previous studies have shown that HIF1 is activated in tissue hypoxia and is rapidly degraded during normal tissue oxygenation [20, 21, 22]. On the other hand, oxygen excess can inhibit the formation of new vessels and even their regression, similar to oxygen-induced retinopathy in premature infants [23].
Immunohistochemical studies have shown that in colorectal cancer lymphatic metastasis is correlated with a high level of HIF-1a [24]. Recently it has been reported that HIF-1a has a crucial role in starting on the chain of reactions that lead to carcinogenesis [25, 26]. This subunit, HIF-1a binds to CXCR4 which promotes angiogenesis as well as hematopoiesis and migration of malignant cells [27, 28, 29]. Recent research suggests that the pro-angiogenic activity of HIF-1a is enhanced by environmental toxins such as cadmium increasing significantly the risk of pulmonary adenocarcinoma [30]. Moreover, in-vitro studies conducted in prostate cancer cells indicate HIF1 controls transcription of miR-182 a factor which subsequently acts in concert with HIF to promote vascular development of prostatic cancer cells. It suggested that this synergic action of these two factors would result in an adaptation to hypoxic stress conditions during prostate cancer growth [31]. In oral squamous cell carcinoma, VEGF-A synthesis and subsequent cancer angiogenesis and development are enhanced by activation of the αvβ3/FAK/c-Src pathway which ultimately would stimulate the final molecular pathway represented by the EGFR/ERK/HIF1-α complex [32]. A detailed evaluation of tissue from smokers with a chronic obstructive pulmonary disease has suggested that an up-regulated HIF-1 may induce lung cancer in a K-ras mutant murine model [33]. All these data indicate that HIFs are a potential therapeutic target.
Recently, based on the HIF structure several antagonists have been developed. Studies conducted in-vitro in human breast cancer cell line (MDA-MB-231) have revealed that analogues of the CJ-3k antagonist have an efficient anti-HIF activity suggesting that they might be effective as anti-cancer pharmacological agents [34]. Also, Qin RS et al. (2018) have successfully used metronomic therapy (low dose – high frequency) with Vinorelbine (NVB) administered with a known angiogenesis inhibitor, Endostar, to treat animal subjects with non-small cell lung cancer [35]. In the same study, they observed a significant decrease of HIF-1 as well as VEGF and CD31 associated with a decrease in tumour development as assessed by micro fluorine-18-deoxyglucose PET/computed tomography (18F-FDG PET/CT) [35]. However, the authors did not report a reduction in cancer-associated angiogenesis. This observation would suggest that while VEGF/HIF-1 are important for tumour growth the above mentioned metronomic therapy might have a direct anti-tumoral effect unrelated to the theoretical anti-angiogenic potential of the proposed therapy. Alternatively, one could speculate that the anti-tumoral effect is much more rapid than the anti-angiogenic effect of Vinorelbine combined with Endostar [35].
c. Matrix metalloproteinases (MMPs)
Angiogenesis requires degradation of the vascular basement membrane and extracellular matrix remodelling to allow endothelial cells to migrate and invade the surrounding tissues. Matrix metalloproteinases (MMPs) represent a group of several proteolytic enzymes which metabolize various extracellular matrix components including the basement membranes and the extracellular matrix facilitating migration of endothelial cells into surrounding regions [36].
Recent studies conducted in a murine model of ischemic stroke have reported that increased levels of matrix metalloproteinases-2 (MMP-2) are associated with post-stroke angiogenesis [37]. Increased levels of matrix metalloproteinases have been recorded in patients with traumatic brain injury. In this context, it seems that matrix metalloproteinases are responsible for blood-brain barrier disruption and leakiness [38]. Anyway, matrix metalloproteinases-2 (MMP-2) and matrix metalloproteinases-9 (MMP-9) promote retinal angiogenesis. However, inhibition of both factors by RNA interference (RNAi) stops the expansion of human retinal microvascular endothelial cells [39]. In addition, matrix metalloproteinases might also be involved in the process of diabetic wound healing. ND-36, an inhibitor of MMP-2 and MMP-9 administered in parallel with MMP 14 induces healing of chronic diabetic wounds by decreasing inflammation in association with stimulation of the angiogenesis process which in turn will promote wound epithelization. The same results have been recorded after administration of recombinant MMP8 [40].
However, matrix metalloproteinases-2 (MMP-2) and matrix metalloproteinases-9 (MMP-9) are inducing angiogenesis in cancer-promoting metastatic spread. In vitro studies performed on prostate cancer cell lines have shown that MMP9 overexpression is also associated with increased levels of murine double minute 2 (MDM2). The authors suggest that MDM2 might promote MMP9 driven angiogenesis [41]. Other experiments have indicated that the level of MMPs is modulated by α2β1 integrin in breast cancer and endothelial cells [42]. Webb et al. (2017) have indicated that MMP9 and MMP2 promote the process of vascular metastasis and tumour growth in retinoblastoma, an ocular neoplasm described in children. This suggests that both MMPs are valid therapeutic targets in retinoblastoma patients [43]. Anyway, MMP-2 is associated with a low rate of survival in patients with colorectal cancer. Interestingly, MMPs may be inhibited by naturally occurring inhibitors. Allyl isothiocyanate (AITC) has been detected in a number of vegetables. Its action results in a decrease in the levels of MMP-2, MMP-9, and mitogen-activated protein kinases (MAPK) –p-JNK, p-38 and p-ERK [36]. Recent studies have indicated that matrix metalloproteinases-9 (MMP-9) is controlled by mitochondria-associated reactive oxygen species (mtROS). The mtROC activity is in turn stimulated by NADPH oxidase 4 (NOX 4) and overall cancer metastasis is promoted [44]. However, at the present time, new factors are tested for their anti-MMP action in cancer metastasis. In this context, eupatilin inhibits angiogenesis-mediated in human hepatocellular lines by decreasing the levels not only of MMP-2 but also VEGF and HIF-1 [45].
Also, MMP2 and MMP9 could be involved in the etiopathogenesis of preeclampsia. In a murine model of reduced uterine placental perfusion, it was discovered that the level of both these two factors is considerably reduced compared with normal subjects. Therefore, it was concluded that MMPs may be responsible for a significant reduction in uterine and placental vascularization [46].
Overall, these data would suggest that matrix metalloproteinases would have complex differential effects in various tissues, but at the present time, we do not have efficient pharmacological agents to modulate the activity of MMPs.
d. Angiopoietins/Tie-2
Recent studies suggest that angiopoietins are of paramount importance in normal angiogenesis [47]. This group includes several factors such as Ang-1, Ang-2, Ang-3 and Ang-4. Currently, it is known that Ang-1 which acts upon endothelial cells on Tie-2 which is a receptor tyrosine kinase promoting endothelial cells stabilization and their survival. Woolf et al. (2009) suggest that genetically Ang-1-deficient animals die as a result of vascular immaturity, while those subjects deficient in Ang-2 die very shortly after birth, displaying anomalies of retinal vascularization as well as abnormal lymphatics. These results indicate that angiopoietins are crucial for vascular differentiation and maturation [48].
Subsequent to the activation of Tie-2, mesenchymal cells are recruited to induce pericyte attachment and ultimately the formation of "non-permeable" vessels. It was reported that Ang-2 in low doses inhibits the activation of Tie-2 but high concentrations of Ang-2 would promote the activation of Tie-2 [49]. Furthermore, Felcht et al. (2012) have confirmed that Ang-2 binds to Tie-2 and behaves as a negative modulator of Ang-1/Tie-2 binding [50]. Therefore, it is considered that Ang-2 is an Ang-1 antagonist which prevents pericyte attachment but induces angiogenesis in the presence of VEGF-A [51]. In patients with diabetic retinopathy, in concert with VEGF, Ang-2 and Tie-2 promote neovascular development while breast hyperplasia is associated with an Ang-2 pro-angiogenic response [52, 53]. Further experiments conducted in a mice model of myocardial infarction have shown that myocardial ischemia promotes an increase of Ang-2 and Tie-2. However if Ang-1 level is higher than Ang-2 the size of the myocardial ischemic lesion is smaller than in cases in which Ang-2 is elevated over Ang-1 [54].
In healing wounds, Ang-1, Ang-2 and Tie-2 are identified at the level of myofibroblastic cells. Their level correlates positively with VEGF and new vessels formation [55]. In some cancers such as glioma, angiogenesis could be promoted subsequent to Ang-2 binding to alpha(v)beta(1) integrin on endothelial cells and activation of focal adhesion kinase signaling pathway (FAK) as well as an over-expression of matrix metalloproteinase 2 (MMP2) which is a pro-angiogenic factor [56] (Figure 3).
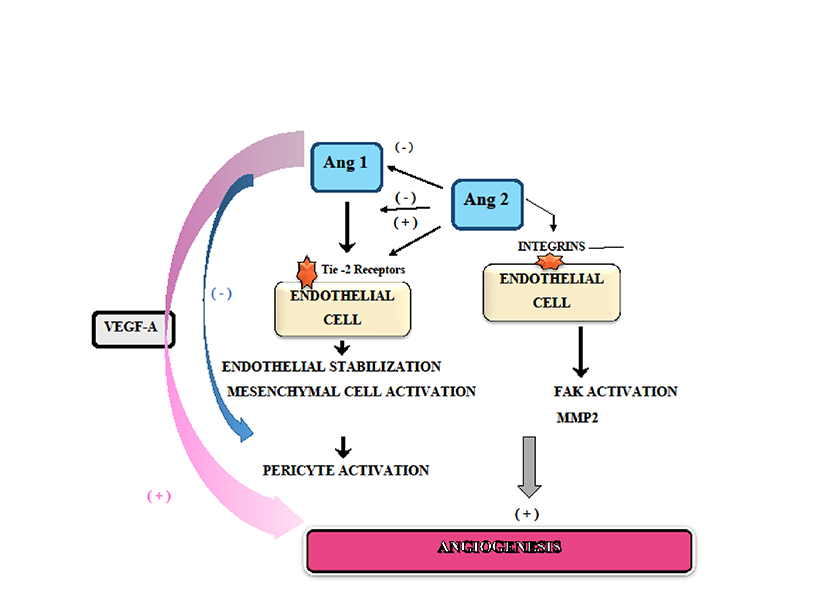
Figure 3
Figure 3 caption
Angiogenesis and Angiopoietins/Tie-2 system
The Ang-1 / Tie-2 interaction promote the development of metastases in cases of neuroendocrine tumours of pancreas and stomach attracting in close proximity to a tumour a population of monocytic cells expressing Tie-2. It seems that these cells enhance both tumour-associated angiogenesis and regional inflammation [57]. However, recent in vitro research using mesothelioma cells over-expressing Ang-1 and Ang-2 transplanted in rats have revealed that low Ang-1 level is characteristic for the epithelioid mesothelioma type while tumours characterized by both high levels of Ang-1 and Ang-2 are associated with vascularized lesions [58].
Interestingly, factors targeting Ang-2 in parallel with VEGF such as ANG2 have been recently developed. ANG2 reduces vascularization in melanoma, pancreatic and breast cancer while inducing tumour necrosis via a CTL-dependent mechanism promoting perivascular deposition of CD8+ cytotoxic T lymphocytes (CTLs) which express interferon-γ (IFNγ) as well as programmed cell death ligand 1 (PD-L1) in a tumour's endothelial cells. This suggests that the anti-VEGF-a and anti-Ang2 action of ANG2 could be further enhanced by PD-1 inhibition [59].
e. Notch signalling
This molecular pathway modulates cellular development during embryonic development [60]. In animal models, a deficiency of Notch-1 determines an early embryonic death due to disruption of the angiogenesis process [61]. The Notch family of proteins includes Notch-1, Notch-2, Notch-3, and Notch-4 representing a group of transmembrane heterodynamic receptors that interact with the ligands located in the cell membranes: Jagged1, Jagged2, and delta-like ligands (DII1, DII3, and DII4) [62]. It was suggested that Notch pathway functionality is modulated by factors such as multiple- post-synaptic density protein (MPDZ) by facilitating the interaction between DLL1/4 with Nectin-2 which is located at the adherens location. Inhibition of MPDZ activity is associated with Notch pathway inhibition and angiogenesis. However, the newly formed blood vessels are not functional [63].
Recent studies conducted in a transgenic murine model have revealed that endothelial Notch modulates the synthesis of local lipase as well as other factors such as Fabp4, Angptl4 and CD36 which may be regulating the transport of free fatty acids across the endothelium. It was shown that in mutant animals with non-functional Notch signalling there is a significant accumulation of lipids in plasma [64].
The Dll4/Notch-1 has a crucial role, modulating VEGF-dependent luteal angiogenesis [65]. Notch1 acts on Hey2 and HeyL and modulate myofibroblastic transformation of rat hepatic stellate cells. Also, Notch-1 is expressed by the synovial tissue enhancing VEGF/Ang-2 angiogenesis in inflammatory arthritis in response to hypoxia [66, 67, 68]. In addition, Notch- 1 has a role of paramount importance in pulmonary hypertension, modulating vascular remodelling [69]. Moreover, Notch signalling has a significant role in skeletal development, as well as bone remodelling. Mutations of Notch-2 are associated with Hajdu Cheney syndrome characterized by extensive osteoporosis and fractures. Histological evaluation in these cases has revealed osteopenia and an increased number of osteoclasts. Interestingly, all these abnormalities are corrected by administration of a γ-secretase inhibitor [70].
Recent research investigating the endothelial cells have proved in a mice model that Notch pathways and the process of angiogenesis induced by this pathway is modulated by the SDF-1-CXCR4 pathway and the VEGF-A [71]. Notch pathway is inhibited by DAPT, a γ-secretase inhibitor. This is followed by significant vascularization of endometriotic lesions in C57BL/6 mice. However, the DAPT is not associated with any morphological changes in the endometriotic lesions suggesting that other molecular factors/pathways are implicated in the etiopathogenesis of this lesion [72].
However, a recent clinical study has revealed that Notch3 and Jagged-1 are significantly increased in cases of pituitary adenomas but the actual modulation of a potential molecular chain of events is not yet fully elucidated [73]. Previously, Dufraine et al. (2008) have suggested that Notch ligands might be involved in tumour angiogenesis [74]. Interestingly, blocking DII4 resulted in an increased vascular development associated with a significant reduction in proliferation of tumour cells which is not completely understood Jagged1 in contrast, was described as facilitating the proliferation of tumour endothelium [74]. On another perspective, recent research has indicated that arresting DII4-Ntotch1/Notch 4 interaction in the setting of the ischemic retina is very useful as it induces new capillaries development [75].
All these data would suggest that Notch signalling could represent a therapeutic target in selected cases of chronic inflammatory conditions and cancer.
f. Wnt/b-Catenin
Reis et al. (2012, 2013) suggest that Wnt/b-catenin complex is important for organ differentiation, vascular remodelling and angiogenesis [76, 77]. This complex is also involved in bone and cartilage remodelling, inducing healing of the osseous tissue [78]. Wnt/b-catenin pathway modulates endometrial physiological function as well as carcinogenesis [79]. Recent studies conducted in rats fed with a high-fat diet have suggested the Wnt pathway promotes a sustained production of IL-6, TNF-α, DKK and VEGF which act in concert to stimulate angiogenesis and atheroma plaque [80].
The Wnt/b-catenin pathway has a crucial role in the formation of the blood-brain barrier and negative modulation of cerebral tumoral angiogenesis after inducing platelet-derived growth factor- beta (PDGF-B). Interestingly, in the above-mentioned setting, Notch-1 activates the cascade Wnt/b-catenin-Dll4/Notch-1 inhibiting process of cancer associated angiogenesis [76, 77]. Recent reports indicate that angiogenesis and vascular differentiation may be modulated by the Wnt ligands, more specifically by the Wnt 7a-Wnt 7b complex composed of MMP inhibitor Reck and an adhesion factor, Gpr124. They modulate not only brain angiogenesis but also dorsal root ganglia neurogenesis [81]. Therefore, it is not surprising that in this context, beta-catenin deletion in endothelial cells leads to developmental angiogenic defects in the central nervous system [82, 83, 84].
However, Wnt pathway seems to promote the process of cancer metastasis and development in other types of` cancer. Abnormalities related to the Wnt /b-catenin pathway have been described in cases of pancreatic cancer resistant to chemotherapy [85]. The Wnt-ligand is a secreted glycoprotein that binds to Frizzled receptors, which triggers a molecular "cascade" resulting in the displacement of the multifunctional kinase Glycogen Synthase Kinase (GSK)-3β from the Adenomatous Polyposis Coli (APC)/Axin/GSK-3β-complex. However, if Wnt is blocked, β-catenin is degraded by the APC/Axin/GSK-3β-complex. Also, phosphorylation of β-catenin by CK1 and GSK-3β leads to its degradation through the β-TrCP/SKP complex. This complex has been revealed to be very important in lung carcinogenesis [86]. In addition, Linke et al. (2017) have indicated that in cases of Hodgkin’s lymphoma, Wnt activates malignant cell chemotaxis towards the blood vessels as well as the local process of angiogenesis [87].
Interestingly, the root of Scutellaria baicalensis Georgi contains a flavonoid named Wogonoside which in cases of breast cancer has a strong anti-angiogenic action by inhibiting the Wnt/β-catenin pathway. In fact, Wogonoside increases the Axin/Glycogen Synthase Kinase (GSK)-3β complex and induces phosphorylation of β-catenin subsequent to an intracellular decrease of Wnt3a [88]. In addition, Park et al. (2011) have identified a pigment epithelium-derived factor (PEDF) which would inhibit Wnt/b-catenin [89]. All these data would indicate without doubt that the Wnt/b-catenin molecular pathway represents a potential therapeutic target of significant clinical importance.
g. Cyclin A1- CCNA1
Cyclin A1-CCNA1 represents a 60 kDa protein which acts as a cell-cycle modulator synergic with cyclin-dependent kinases (CDKs), activating complex enzymes [90]. It also acts during meiosis and during the progression of the cellular cycle from G1 to S in somatic cells [91]. Recent studies have reported that the activity of CCNA1 is dependent on S100A6, a calcium binding protein that activates the endothelial cell-cycle senescence and progression. A decrease in this factor induces a significant decrease in CCNA1 and abnormalities of cell-cycle progression [92]. Bauer et al. (2016) have reported that CCNA1 represents a valid target for heme oxygenase -1 (HO-1). Overall, this interaction promotes enhanced angiogenesis [93]. Various cancers related to epigenetic changes are characterized by increased levels of methylated CCNA1. Testicular embryonal carcinomas and teratomas present with the highest levels of this factor. A hyper-methylated precursor of CCNA1 is recovered in the urine of vesical bladder cancer patients [94, 95]. In addition, increased CCNA1 levels have been reported in head and neck squamous cell carcinomas, as well as in melanomas and hepatocellular carcinomas [96, 97]. More importantly, it seems that CCNA1 acts in concert with VEGF in breast, prostate and ovarian carcinogenesis. A high level of CCNA1 suggests an increased proliferative risk, while an increased VEGFR2 expression indicates an advanced stage of cancer [98]. In prostate cancer, CCNA1 promotes invasiveness by acting upon androgen receptors and modulating MMP and VEGF expression [99]. Normal breast tissue displays low levels of CCNA1 while breast cancer tissue reveals both an increase in CCNA1 and VEGF. Remarkably, CCNA1 binds estrogen receptors (ER) suggesting that breast cancer progression results from multiple synergic interactions between various molecular pathways including modulators of angiogenesis such as VEGF but also cell cycle factors such as CCNA1 [100] (Figure 4).
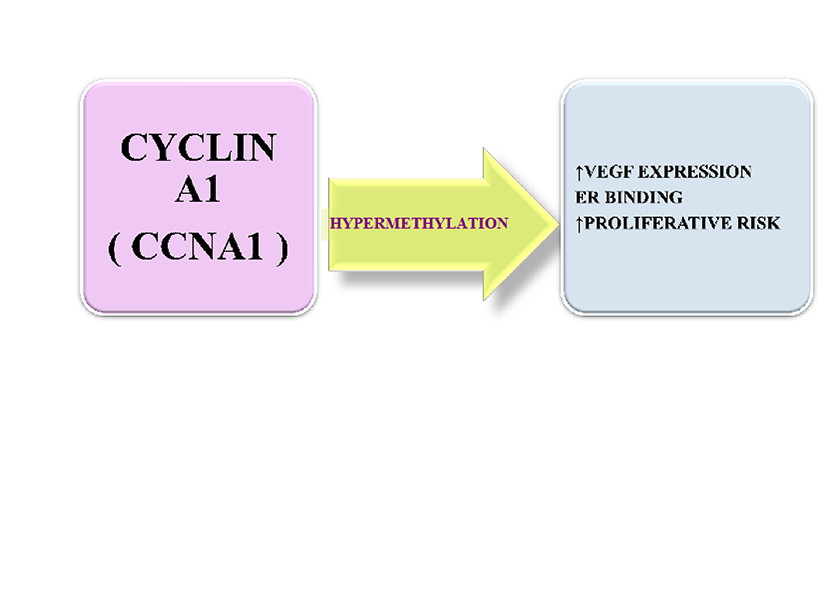
Figure 4
Figure 4 caption
Cyclin A1: mechanism of action
h. Follicle stimulating hormone (FSH)
Radu et al. (2010) have shown that FSH is present in the vascular network associated with a wide range of cancers such as breast, colon, pancreas, urinary bladder, kidney, lung, liver, stomach, testis and ovary. Also, FSH receptors have been identified in both benign and malignant tumours of adrenal gland [101, 102].
FSH receptors are also seen in metastases [103]. Until recently it was accepted that there are no FSH receptors in the normal tissues except in the ovarian and testicular parenchyma, more exactly in the granulosa and the Sertoli cell [104]. However, recent clinical studies have reported that FSH receptors are also located in the endothelial cells of the cervix, endometrium, myometrium including in the endometrial and cervical glands and myometrial muscle fibres. In addition, in pregnant women, FSH receptors are seen on the endothelial cells associated with umbilical cord and in both the fetal and maternal placenta. However, the role of these receptors in these locations is not known [105].
Regarding the pro-angiogenic role of FSH, it was reported that binding to its receptor in the ovarian granulosa cells determines an up-surge of hypoxia-inducible factor 1 alpha (HIF-1 alpha), which subsequently determines an increase of vascular endothelial growth factor (VEGF) which is known to promote a rapid angiogenesis which has a significant role in further follicular development [106, 107]. Experimentally, it has been proven that FSH treatment during ovarian cryopreservation promotes ovarian survival by neo-angiogenesis subsequent to a sustained increase in VEGF/ VEGFR2 [108]. In avian ovarian follicles, FSH induces a robust VEGF-A angiogenesis after prior induction of Annexin A2 (ANXA2) which is a calcium-dependent, phospholipid-binding protein [109]. Studies completed recently using ovarian epithelial cancer cells indicated that FSH increases the VEGF expression also via production of reactive oxygen species (ROS) which in a second stage would turn on the Nrf2 pathway [110]. However, these findings have not been verified in a clinical setting but it is suggested that FSH exerts its positive effect on VEGF over-expression and associated angiogenesis by interacting with a variety of factors and molecular pathways. Overall, these data suggest that FSH could be a significant therapeutic target.
i. PTEN/PI3K–AKT complex
In general, PI3K–AKT induces angiogenesis while overproduction of PTEN inhibits angiogenesis in the chicken embryo [111]. It seems that stimulation of PI3K/AKT pathway and simultaneous suppression of PTEN has a beneficial effect protecting the heart from reperfusion injury [112].
PTEN (phosphatase and tensin homolog deleted on chromosome 10), an antagonist of PI3K-AKT, is a tumour suppressor, which is altered in several human cancers [113]. In addition, PTEN represents a target for miR-19a, a member of the microRNA clusters which modulates angiogenesis. Down-regulation of PTEN by miR-19a is followed by activation of AKT/GSK pathway and glycogen synthesis in hepatic cells [114]. Recent scientific reports suggest that in endothelial cells, PTEN is down-regulated by urokinase receptor (uPAR/CD87). Interestingly, PTEN down-regulation is associated with an increased density of newly formed blood vessels [115]. However, studies conducted in a PTEN deficient mouse model of prostate cancer have revealed that KLF5, a recently described transcriptional factor, induces cancer development subsequent to an increase in HIF1α and inhibition of PI3K-AKT pathway [116].
The PI3K-AKT complex has a crucial role in modulating cell proliferation and growth as well as cellular survival initiated by other growth factors. Mice deficient in p110a catalytic subunit of PI3K have more vascular defects including dilated cerebral vessels, anomalies endocardium development as well as a significant decrease in Tie-2 protein level. More importantly, in p110g deficient mice, the vascular permeability induced by RAS and VEGF is significantly decreased. This suggests that PI3K has a crucial role in maintaining an adequate vascular permeability [117].
Some authors believe that PI3K is one of the most important molecular pathways implicated in carcinogenesis [118]. Erdreich-Epstein et al. (2016) have reported in a murine model of neuroblastoma that deletion of one allele of PTEN stimulates malignant development. This supports further assessment of factors targeting this pathway such as SF1126 as chemotherapeutic agents [119].
Remarkably, PI3K is sufficient by itself to induce angiogenesis partly by increasing the expression of HIF-1 and VEGF. A number of studies have demonstrated a modulation by PI3KofHIF-1andVEGFexpression in vascular endothelial cells, several types of neoplastic cells, smooth muscle cells, osteoblasts and mast cells [120]. However, PI3K/AKT interacts with other molecular factors and pathways to promote angiogenesis. Immuno-histochemical studies conducted in patients with gastric cancer have indicated that PI3K/AKT is simultaneously activated with mTOR molecular pathway while the PTEN is downregulated [121].
j. B-Cell-specific Moloney Murine Leukemia Virus Integration Site 1 (Bmi-1)
B-cell-specific Moloney murine leukaemia virus Integration site1 (Bmi-1) represents a member of gene polycomb family. It was reported to have a significant etiopathogenic role in breast cancer, acute myeloid leukaemia as well as colorectal, and lung cancer, ovarian and nasopharyngeal tumours. In all these cancers, Bmi-1promotes malignant cell proliferation and progression inhibiting apoptosis and cellular senescence [122, 123, 124].
Bmi-1 interacts with numerous molecular pathways implicated in cancer such as inhibition of p16/Rb and p19ARF/MDM2/p53 as well as Hedgehog pathway. It stabilizes the snail factor which is a transcription repressor which subsequently promotes epithelial-mesenchymal transition through modulation of PI3K/AKT/GSK-3b [125, 126]. Bmi-1 promotes hepatic angiogenesis and modulates the NF-kB/VEGF-C which is very important in cancer growth and development, particularly in gliomas. Other studies have suggested that aggressive gliomas are determined after activation by Bmi-1 of NF-kappaB/MMP-9 molecular pathway [127, 128]. In head and neck squamous cell carcinomas, cisplatin administration promotes Bmi-1 synthesis and cancer stem cells formation. This could represent a potential mechanism of cancer resistance to cisplatin therapy [129]. These results would suggest that Bmi-1 could represent a potential therapeutic target in selected cases of cancer.
k. Galectins
Recent research suggests that Galectins are a group of pro-angiogenic molecules with a significant role in a multitude of pathological conditions including cancer and pre-eclampsia [130]. Therefore, numerous studies have been conducted to assess the potential of these molecules as therapeutic targets. In this context, Galectin-3 is a lectin which binds to b-galactosidase and displays three distinct structural domains: an NH2-phosphorylation site, a collagen repeating part and a terminal COOH domain for binding carbohydrates. Galectin-3 has a role of paramount importance in embryonic growth and development, apoptosis, cellular differentiation and proliferation, and modulation of the immunological system. It is expressed by macrophages, eosinophils, and neutrophils and mastocytes. In addition, it is seen in gastrointestinal and respiratory epithelium, kidneys and sensory neurons [131].
Tissue remodelling after neural or heart ischemia is characterized by increased expression of galectin-3 [132]. Recent research suggests that galectin-3 is a pro-angiogenic factor promoting the action of VEGF and b-FGF [133]. Dos Santos et al. (2017) have indicated that galectin-3 in concert with hypoxic conditions stimulates new blood vessel formation via activation of the JAG1/Notch-1 molecular pathway [134]. Also, Etulain et al. (2014) have demonstrated that Galectin-1, 3 and 8 exert their pro-angiogenic role via VEGF release from human platelets promoting in vitro tubular formation and endothelial cell proliferation [135].
Interestingly, migration of type M2 cancer macrophages is taking place according to a galectin-3 gradient and increased expression of this protein is correlated with malignant angiogenesis, tumour growth and aggressiveness [136]. Research conducted in a melanoma cell line has indicated that in transforming growth factor beta 1 (TGFβ1) activated macrophages, tumoral angiogenesis is impaired by inhibition of Galectin-3 pathway [137].
At the present time, galectin-3 explains progression in breast, prostate and pancreatic cancers, melanoma, follicular thyroid tumours and neuroblastoma [138, 139, 140, 141, 142, 143]. Also, in oral squamous cell carcinoma, galectin-3 in parallel with another lectin named galectin-1, promotes angiogenesis, cell proliferation and survival [144].
Park et al. (2015) have shown that in cultures of osteosarcoma cells, inhibition of galectin-3 is followed by down-regulation of VEGF, IL-8, IL-6, phospho-Stat3, MMP2/9 and MCP-1 as well as an inhibition of several molecular pathways such as Wnt/beta-catenin, FAK, ERK1/2, PI3K/AKT, Src and Lyn. All these are translated into a significant decrease in cell migration and invasiveness [145]. In Her-2 negative breast cancer cells, galectin-3 in concert with Annexin-A 2 modulates the epidermal growth factor receptor pathway which promotes cancer development [146]. In breast tumour development, hypoxia significantly increases production of galectin-3 which seems to be responsible for an aggressive phenotype while in concert with senescent cell-derived growth factors, galectin-3 induces colorectal cancer cells growth [147, 148].
l. Advanced Glycation End Products (AGEs)/ Receptors for Advanced Glycation End-products (RAGEs)
Hoffmann et al. (2002) have observed that in the eye, Advanced Glycation End Products (AGEs) not only that increase in newly formed blood vessels but also promote proliferation of Müller cells, retinal pigment epithelial cells, and choroidal endothelial cells [149]. AGEs are proteins or lipids modified by non-enzymatic glycation and oxidation processes. These appear in diabetic chronic hyperglycemia due to oxidative stress. AGEs act upon specific receptors named receptors for advanced glycation end products (RAGEs). These are seen in monocytes and macrophages, smooth muscle fibers, lymphocytes and endothelial cells [150]. AGEs–RAGEs interaction, triggered by the activation of an inflammatory process potentiated by NF-kB stimulates the accumulation of extra-cellular matrix accelerating the formation of atherosclerotic plaques and cardiac fibrosis [151]. Recently published studies have revealed that suppression of RAGE improves the angiogenic response to ischemia. The authors explain the observed effect by a significant increase in VEGF [152]. Interestingly, this VEGF related mechanism has also been described in human synoviocytes [153]. Recent studies suggest that RAGEs could be involved in carcinogenesis by modulating the VEGFR2 and increasing vascularization in renal cell cancer patients. RAGEs also correlate with a reduced survival as well as decreased metastasis-free evolution. All these suggest that RAGEs could be a prognostic marker in patients with renal cell carcinoma [154]. Additional experimental studies have revealed that interaction AGEs-RAGEs in glioma-associated microglia promotes angiogenesis [153].
m. HMGB1
The HMGB1 protein acts upon several receptors TLR4 (Toll-like receptor 4), RAGE (receptors for advanced glycation end-products), CD24 and TIM-3 and modulates replication, transcription, recombination, and repair of DNA [155, 156]. In vitro studies have shown that HMGB1 promotes endothelial cell migration and sprouting of newly formed blood vessels in a dose-dependent manner [157]. HMGB1 initiates expression of Ang-2 and angiogenesis in skeletal muscle in response to tissue hypoxia [158]. HMGB1 interacts with RAGEs and stimulates the production of pro-angiogenic and pro-inflammatory factors [159].
Also, as results of its interaction with RAGEs, HMGB1 facilitates the pro-tumoral action of type 2 macrophage phenotype induced by IL4/Il-13, including angiogenesis [160]. After intra-cerebral haemorrhage, HMGB1 acts also upon RAGEs to promote angiogenesis via up-regulation of VEGF [161]. Studies performed in a murine model of rheumatoid arthritis have indicated HMGB1 would exert its pro-angiogenic effect in synovium via stimulation of HIF-1α. In this context, therapy with anti-HMB1 antibody would prevent the development of new blood vessels as well as synovial inflammation while inducing a significant decrease in HIF-1α [162].
Interestingly, in the cancer setting, HMGB1 interacts with mitogen-activated protein kinase (MAPK)-NF-kB and mTOR molecular pathways but it seems that HMGB1 has a dual effect [155, 163]. On one side it stimulates cancer-associated angiogenesis while promoting the anti-neoplastic effects of T lymphocytes [164, 165, 166]. In acute myeloid leukaemia, HMGB1 promotes malignant development and progression via increased VEGF secretion which is dependent on the functionality of Tim-3 immune receptor [167].
n. Leptin
Leptin is a factor or an “adipokine” secreted by the adipose tissue which acts at the central nervous level to regulate satiety and appetite. It also interferes with endothelial dysfunction which is a risk factor for atherosclerosis [168]. Leptin levels correlate with adipose tissue size and body weight modulating bone physiology, immunological function and reproduction. More importantly, leptin induces the formation of new blood vessels [169]. Recent research suggests that leptin levels promote proliferation and differentiation of angiogenic endothelial cells in vitro and in vivo [170]. Leptin modulates the process of angiogenesis in skeletal muscles acting as a paracrine factor via platelet-derived growth factor receptor α (PDGFR α) and platelet-derived growth factor receptor β (PDGFR β) positive endothelial cells [171].
Also, leptin seems to have a significant role in the embryological development of the cardiovascular system. The hypoplastic left heart syndrome is characterized by leptin dysregulation. Since leptin has a pro-angiogenic effect, recent studies suggest that high levels of leptin recorded in subjects with hypoplastic left syndrome might be a natural attempt at correcting morphological abnormalities of the cardiovascular system [172]. Leptin promotes skin wound healing. Experimental studies conducted in a mouse model have indicated that topically administered leptin acts upon leptin receptor located in the skin to facilitate the differentiation and functionality of keratinocytes and local angiogenesis which would promote wound healing [173]. Moreover, leptin is capable of promoting expression in various tissues of a wide range of pro-angiogenic factors including the most powerful pro-angiogenic mediator, VEGF and its receptor VEGFR2. It also promotes the synthesis of IL1, Notch, MMPs, TIMP and integrins [174].
Leptin has been linked to the development of breast cancer [175]. Interestingly, the leptin receptor and estrogen receptor are co-expressed in breast cancer suggesting that leptin may play a significant role in the development of estrogen-dependent breast cancer [176]. A clinical study conducted in a cohort of patients with colorectal carcinoma has found that almost 80% of cases are characterized by the presence of leptin receptors in the lesion. This correlates positively with cancer-related angiogenesis and cellular proliferation [177]. The authors suggest that leptin profile could be used to select those patients which are at high risk of developing an aggressive form of colorectal cancer. Sobrinho Santos et al. (2017) have demonstrated in an animal model of oral squamous cell carcinoma that the levels of leptin and its receptor are increased in parallel with HIF-1. However, administration of leptin induces a significant increase in MMP9 and 2, E-cadherin and mir-10 [178]. All of these are known to promote angiogenesis and tumour development.
o. Leucine-Rich Alpha-2-Glycoprotein 1 (LRG1)
Leucine-rich alpha-2-glycoprotein 1 (LRG1) is expressed during the differentiation of granulocytes and it is detected in human neutrophils and in their precursor cells, but not in peripheral mononuclear cells. In patients with non-small cell lung cancer, an increased level of LRG1 was reported in plasma and urine [179, 180]. Increased LRG1 expression is also observed in inflammatory bowel disease correlated with disease activity [181]. LRG1 stimulates angiogenesis via the TGF-b pathway and could have a significant role in cardiac remodelling preventing or attenuating the hemodynamic effects of heart failure [182]. Studies conducted in an animal model have revealed that LRG1 is co-expressed in retinal vasculature along with transforming growth factor-b1 (TGF-b1). In this context, LGR1 induces cellular proliferation and angiogenesis. It seems that this molecular pathway is switched on by the binding of LRG1 on endoglin which is the TGF-b1 receptor. This results in activation of Smad1/5/8 which ultimately results in new blood vessels formation. Moreover, administration of an LRG1 antibody results in the inhibition of angiogenesis. Also, subjects without LRG1 display a significant reduction in pathological retinal angiogenesis [183].
p. NADPH Oxidase1
NADPH oxidase (Nox) family protein consists of seven isoforms (Nox1–5, DUOX1–2) which transport oxygen through the cellular membrane. This leads ultimately to a significant reduction in superoxide oxygen. The isoenzyme Nox 1 is expressed by the epithelial cells, pericytes, osteoclasts, smooth muscle cells and endothelial cells [184]. However, in the resting endothelial cells, Nox 1 is expressed at a low level. Remarkably, Nox 1 level increases significantly after the beginning of angiogenesis. Recent studies have mentioned that the synthesis and release of pro-angiogenic factors such as angiopoietin 1 and VEGF have been shown to be dependent on Nox 1 and Nox2. VEGF, as well as its receptors, VEGFR1 and VEGFR2, are over-expressed in the presence of Nox-1 in both tumour tissues and cell cultures [185, 186]. Vara et al. (2018) have demonstrated that 2-Deoxyribose-1-Phosphate activates NADPH Oxidase 2 which subsequently stimulates angiogenesis via activation of nuclear factor kappa B (NF-κB) [187].
In-vitro studies have shown that Nox1 modulates apoptosis and morphogenesis of sinusoidal endothelial cells suggests that it might have a significant role in the process of vascular development [184]). Previous experiments have revealed that Nox1 is involved in RAS oncogenic transformation by promoting over-expression of VEGF. Small interference NOX1 RNA inhibits the extracellular signal-regulated kinase (ERK) dependent activation of VEGF promoter (Sp1). Overall, there is a significant decrease in angiogenesis. These effects have been observed in both K-ras transformed kidney cells as well as in human colorectal cancer cells [188]. In a culture of K-Ras-activated kidney cells, transfection of Nox1 small interference RNA (siRNA) decreases angiogenesis and cell migration by blocking NF-kB dependent matrix metalloproteinase-9 (MMP-9) [189]. Therefore, it is accepted that Nox1 promotes directly the process of angiogenesis. Moreover, increased expression of Nox1 has been reported in clinical cases of colorectal cancer and a recent study has reported that specific inhibitors for Nox4 and Nox2 prevent the development of vascular tumours [184, 186].
q. Urotensin II (UII)
Ross et al. (2010) reported that urotensin II(UII) represents a cyclic peptide with a similar structure as somatostatin which binds a special class of G-coupled receptors namely GPR14 or urotensin receptor [190]. Urotensin II (UII) is expressed in the cardiovascular and respiratory, renal and central nervous system [191]. Recent scientific data suggest that urotensin II (UII) maintains endothelial cell homeostasis and functionality. At this level, it activates via p38 the extracellular signal-regulated kinase as well as mitogen-activated protein kinase molecular pathways to synthesize IL-8 stimulating angiogenesis. Also, the activation of these molecular pathways would also result in cellular proliferation and endothelial cell survival [192]. However, subsequent to its binding to GPR14 receptors, UII activates Nox2 and generates reactive oxygen species (ROS). Then a significant increase in HIF-1a is recorded which further stimulates reactive oxygen species (ROS) production and a major increase in new blood vessels formation via activation of Nox 2 [193].
Molecular evaluation of multiform glioblastoma has indicated that this tumour overexpresses urotensin II (UII) and its receptor. Subsequent to UII interaction with its receptors a molecular chain of events culminating with glioma cell migration and adhesion, is activated. This urotensinergic system, which seems to include the G13/Rho/rho kinase pathway and partially the Gi/o/PI3K molecular complex, is noted in glioma tissue resected from patients as well as biopsy tissue and culture cell lines [194]. In breast cancer, urotensin II (UII) promotes cellular proliferation as well as cancer-related angiogenesis. Moreover, the Thr21Met polymorphism in of the urotensin II (UII) gene, the UTS2 identifies a group of patients at risk of developing breast cancer [195].
r. Orai-1
The gene labelled Orai-1 encodes a calcium-membrane channel subunit namely CRAC (Ca2+release-activated Ca2+) which is activated when calcium deposits are low. These types of calcium channels represent the major route for the influx of calcium in T lymphocytes, modulating their immunological response and subsequently activating the transcription factor NFAT [196, 197]. It was claimed by some authors that increased activation of Orai-1 may represent the molecular mechanism which explains the abnormal control of intracellular calcium described in essential hypertension [198]. The Orai-1 channels enable the cells of the retinal pigment epithelium to produce repetitive and/or sustained Ca²+ signals necessary for various important functions such as transepithelial transport of various substances, growth factor secretion and phagocytosis [199]. In addition, Orai-1 could have a significant etiopathogenic role in diabetic erectile dysfunction [200].
Smooth muscle cells express Orai-1. However, a congenital deficiency of Orai-1 gene leads to myopathies and chronic lung disease due to bronchial smooth muscle damage. As calcium input is essential in tooth development an Orai-1 deficiency such as in anhidrotic ectodermal dysplasia is associated with anomalies of the enamel development. All of these are described within the setting of reduced T lymphocyte activation and cytokine production. However, the vascular structures in these patients are apparently normal which suggests that Orai-1 is not essential for angiogenesis [201]. Anyway, these results have been contradicted by other studies that have found out that Orai-1 is important in the formation of endothelial tubules, but its functionality is dependent on a prior VEGF-activated Ca2+ entry [202]. Moccia et al. (2013) have shown that in early cancer angiogenesis, Orai-1has a positive role facilitating through the respective channel the calcium influx which stimulates the mobilization of endothelial cells and subsequent angiogenesis [203].
In conclusion, modulation of angiogenesis is possible and new factors have shown promising results in experiments conducted in vitro and in vivo. Often times the authors record puzzling effects such as the anti-tumoral effect of combination Vinorelbine and an anti-angiogenic agent such as Endostar in subjects with non-small cell lung carcinoma in which cancer-associated angiogenesis was not affected [35]. Other studies have revealed important physio-pathological mechanisms which must be further investigated such as the role of Tie-2 in impermeabilization of blood vessels [49]. Other studies have shown that angiogenesis is important in a variety of pathological conditions and theoretically new pharmacological agents could be developed to prevent or treat skeletal malformations, pre-term labour, diabetes and many others. While some of the factors presented above may be acting in parallel, their molecular mechanisms of co-activation and feedback are not yet fully understood. Therefore, more experiments are needed in order to develop efficient pharmacological agents that could effectively modulate angiogenesis in various pathological settings.
Original submitted files for images
Below are the links to the authors’ original submitted files for images.
Original image file for Figure 1
Original image file for Figure 2
Original image file for Figure 3
Original image file for Figure 4
References
- Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73-91.
- Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. 1997 Nov;173(2):206-10.
- Vascular mural cells in healing canine myocardial infarcts. J Histochem Cytochem. 2004 Aug;52(8):1019-1029.
- A plasticity window for blood vessel remodeling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development. 1998 May;125(9):1591-1598.
- Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997 Feb 14;275:964-967.
- uPAR - a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002 Dec;3:932-943.
- Excessive glycation of pericapillary pericytic matrix components is an essential mechanism of arterial hypertension development in hyperglycemia. Bull Exp Biol Med. 2011 Mar;1153(150):572-575.
- Molecular regulation of vessel maturation. Nat Med. 2003 Jun;9:685-693.
- SDF-1 activity on microvascular endothelial cells: consequences on angiogenesis in in vitro and in vivo models. Thromb Res. 2000 Sep 15;99(6):587-94.
- Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Can Res. 2000 Jan;6(1):102-11.
- Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J Exp Med. 2007 Mar 19;204(3):605-18.
- The SDF-1–CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. 2007 Jul;28(7):299-307.
- Angiogenic effects of stromal cell-derived factor-1 (SDF-1/CXCL12) variants in vitro and the in vivo expressions of CXCL12 variants and CXCR4 in human critical leg ischemia. J Vasc Surg. 2010 Mar;51(3):689-99.
- SDF-1 restores angiogenesis synergistically with VEGF upon LDL exposure despite CXCR4 internalization and degradation. Cardiovasc Res. 2013 Dec 1;100(3):481-491.
- CXCL12 enhances angiogenesis through CXCR7 activation in human umbilical vein endothelial cells. Sci Rep. 2017 Aug 15;7(1):8289-.
- The SDF-1/CXCR4 axis utilizes mTOR signal transduction to promote angiogenesis. Pathology. 2014 Apr.:278.8-.
- Stromal cell-derived factor-1 promoted angiogenesis and inflammatory cell infiltration in aneurysm walls. J Neurosurg. 2014 Jan;120(1):73-86.
- Stromal-Cell-Derived Factor (SDF) 1-Alpha Overexpression Promotes Bone Regeneration by Osteogenesis and Angiogenesis in Osteonecrosis of the Femoral Head. Cell Physiol Biochem. 2018;46:2561-2575.
- Cerebral Angioplasticity: The Anatomical Contribution to Ensuring Appropriate Oxygen Transport to Brain. Adv Exp Med Biol. 2018;1072:3-6.
- Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992 Oct 29.
- Hypoxic stabilisation of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem. 1998 Mar;273:6417-6423.
- Endothelial PAS domain protein 1 gene promotes angiogenesis through the transactivation of both vascular endothelial growth factor and its receptor, Flt-1. Circ Res. 2004 Jul;95:146-153.
- A lower oxygen-saturation target decreases retinopathy of prematurity but increases mortality in premature infants. J Pediatr. 2013 Nov;163:1528-1529.
- Clinicopathologic significance of HIF-1a, CXCR4, and VEGF expression in colon cancer. Clin Dev Immunol. 2010.:537531-.
- The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008 Apr;15(4):678-685.
- Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev Mol Med. 2009 Aug 27;11:e26-.
- CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood. 2006 Mar1;107(5):1761-1767.
- Clinical importance and therapeutic implications of the pivotal CXCL12-CXCR4 (chemokine ligand-receptor) interaction in cancer cell migration. Tumor Biol. 2007;28(3):123-131.
- Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004 Aug;10(8):858-864.
- Low-Dose Cadmium Upregulates VEGF Expression in Lung Adenocarcinoma Cells. Int J Environ Res Public Health. 2015 Aug 28;12(9):10508-21.
- Hypoxia-inducible miR-182 enhances HIF1α signaling via targeting PHD2 and FIH1 in prostate cancer. Sci Rep. 2015 Jul 24;5:12495-.
- WISP-1 a novel angiogenic regulator of the CCN family promotes oral squamous cell carcinoma angiogenesis through VEGF-A expression. Oncotarget. 2015 Feb 28;6(6):4239-52.
- COPD-Type lung inflammation promotes K-ras mutant lung cancer through epithelial HIF-1α mediated tumor angiogenesis and proliferation. Oncotarget. 2018 Aug 31;9(68):32972-32983.
- Design, Synthesis and Biological Evaluation of Novel HIF1α Inhibitors. Anticancer Res. 2015 Jul;35(7):3849-59.
- Enhanced antitumor and anti-angiogenic effects of metronomic Vinorelbine combined with Endostar on Lewis lung carcinoma. BMC Cancer. 2018 Oct 11;18(1):967-.
- Allyl isothiocyanate inhibits cell metastasis through suppression of the MAPK pathways in epidermal growth factor-stimulated HT29 human colorectal adenocarcinoma cells. Oncol Rep. 2014 Jan;31(1):189-196.
- Effects of Mesenchymal Stem Cell Treatment on the Expression of Matrix Metalloproteinases and Angiogenesis during Ischemic Stroke Recovery. PLoS One. 2015 Dec 4;10(12):e0144218-.
- Role of Matrix Metalloproteinases in the Pathogenesis of Traumatic Brain Injury. Mol Neurobiol. 2015 Nov 5.
- Silencing Matrix Metalloproteinases 9 and 2 Inhibits Human Retinal Microvascular Endothelial Cell Invasion and Migration. Ophthalmic Res. 2015 Nov 28;55(2):70-75.
- Acceleration of diabetic wound healing using a novel protease-anti-protease combination therapy. Proc Natl Acad Sci U S A. 2015 Nov 23.:201517847-.
- MDM2 Overexpression Modulates the Angiogenesis-Related Gene Expression Profile of Prostate Cancer Cells. Cells. 2018 May 10;7(5):E41-.
- Alternagin-C binding to α2β1 integrin controls matrix metalloprotease-9 and matrix metalloprotease-2 in breast tumor cells and endothelial cells. J Venom Anim Toxins Incl Trop Dis. 2018 Apr 25;24:13-.
- Inhibition of MMP-2 and MMP-9 decreases cellular migration, and angiogenesis in vitro models of retinoblastoma. BMC Cancer. 2017 Jun 20;17(1):434-.
- A mitochondrial ROS pathway controls matrix metalloproteinase 9 levels and invasive properties in RAS-activated cancer cells. FEBS J. 2018 Oct 3.
- Eupatilin inhibits angiogenesis-mediated human hepatocellular metastasis by reducing MMP-2 and VEGF signaling. Bioorg Med Chem Lett. 2018 Oct 15;28(19):3150-3154.
- Angiogenic imbalance and diminished matrix metalloproteinase-2 and -9 underlie regional decreases in utero-placental vascularization and feto-placental growth in hypertensive pregnancy. Biochem Pharmacol. 2017 Dec 15;146:101-116.
- A continuous model of angiogenesis: initiation, extension, and maturation of new blood vessels modulated by vascular endothelial growth factor, angiopoietins, platelet-derived growth factor-b, and pericytes. Dynamic System Ser B. 2013;18(4):1109-.
- Roles of angiopoietins in kidney development and disease. J Am Soc Nephrol. 2009 Feb;20:239-244.
- Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol. 2009 Apr;29:2011-2022.
- Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest. 2012 Jun;122(6):1991-2005.
- Angiogenesis and chronic kidney disease. Fibrogenesis Tissue Repair. 2010 Aug 5;3:13-.
- Plasma vascular endothelial growth factor, angiopoietin-2, and soluble angiopoietin receptor tie-2 in diabetic retinopathy: effects of laser photocoagulation and angiotensin receptor blockade. Br J Ophthalmol. 2004 Dec;88(12):1543-6.
- Angiopoietins 1 and 2 and Tie-2 receptor expression in human ductal breast disease. Histopathology. 2011 Aug;59(2):256-63.
- Critical role of angiopoietins/Tie-2 in hyperglycemic exacerbation of myocardial infarction and impaired angiogenesis. Am J Physiol Heart Circ Physiol. 2008 Jun;294(6):H2547-57.
- Angiopoietin-1, angiopoietin-2 and Tie-2 receptor expression in human dermal wound repair and scarring. Br J Dermatol. 2010 Nov;163(5):920-7.
- Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the alphavbeta1 integrin and focal adhesion kinase signaling pathway. Cancer Res. 2006 Jan 15;66(2):775-83.
- The association of the angiopoietin/Tie-2 system with the development of metastasis and leukocyte migration in neuroendocrine tumors. Endocr Relat Cancer. 2010 Oct 5;17(4):897-908.
- Role of angiopoietins in mesothelioma progression. Cytokine. 2018 Sep 7;S1043-4666(18):30341-7.
- Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci Transl Med. 2017 Apr 12;9(385):eaak9670-.
- Mechanistic insights into Notch receptor signaling from structural and biochemical studies. Curr Top Dev. 2010;92:31-71.
- Notch signaling in developmental and tumor angiogenesis. Genes Cancer. 2011;2(12):1106-1116.
- The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216-233.
- MPDZ promotes DLL4-induced Notch signaling during angiogenesis. Elife. 2018 Apr 5;7:e32860-.
- Inhibition of Endothelial Notch Signaling Impairs Fatty Acid Transport and Leads to Metabolic and Vascular Remodeling of the Adult Heart. Circulation. 2018 Dec;137(24):2592-2608.
- Delta-like ligand 4 regulates vascular endothelial growth factor receptor 2-driven luteal angiogenesis through induction of a tip/stalk phenotype in proliferating endothelial cells. Fertil Steril. 2013 Dec;100(6):1768-1776.
- Notch-1 mediates hypoxia-induced angiogenesis in rheumatoid arthritis. Arthritis Rheum. 2012 Jul;64(7):2104-2113.
- Notch signalling pathways mediate synovial angiogenesis in response to vascular endothelial growth factor and angiopoietin 2. Ann Rheum Dis. 2013 Jun;72(6):1080-1088.
- Myofibroblastic transformation of rat hepatic stellate cells: the role of Notch signaling and epithelial-mesenchymal transition regulation. Eur Rev Med Pharmacol Sci. 2015 Nov;19(21):4130-8.
- Notch signaling change in pulmonary vascular remodelling in rats with pulmonary hypertension and its implication for therapeutic intervention. PLoS One. 2012;7(12):e51514-.
- Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis and Bone Resorption. J Biol Chem. 2016 Jan 22;291(4):1538-51.
- Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat Cell Biol. 2017 Aug;19(8):915-927.
- Notch signaling controls sprouting angiogenesis of endometriotic lesions. Angiogenesis. 2018 Feb;21(1):37-46.
- Expression pattern of the Hedgehog signaling pathway in pituitary adenomas. Neurosci Lett. 2015 Nov 24.S0304-3940(15)30230-5
- Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene. 2008 Sep 1;27:5132-5137.
- The Role of Dll4/Notch Signaling in Normal and Pathological Ocular Angiogenesis: Dll4 Controls Blood Vessel Sprouting and Vessel Remodeling in Normal and Pathological Conditions. J Ophthalmol. 2018 Jul 5;2018:3565292-.
- Wnt signaling in the vasculature. Exp Cell Res. 2013 May 15;319(9):1317-1323.
- Endothelial Wnt/b-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J Exp Med. 2012;209(9):1611-1627.
- Recent research on the growth plate: Mechanisms for growth plate injury repair and potential cell-based therapies for regeneration. J Mol Endocrinol. 2014 Aug;53(1):T45-61.
- Participation of WNT and β-Catenin in Physiological and Pathological Endometrial Changes: Association with Angiogenesis. Biomed Res Int. 2015;2015:854056-.
- The role of Wnt signaling pathway in atherosclerosis and its relationship with angiogenesis. Exp Ther Med. 2018 Sep;16(3):1975-1981.
- Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. Elife. 2015 Jun 8.4
- Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A. 2009 Jan 13;106(2):641-6.
- Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008 Nov 3;183(3):409-17.
- Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008 Nov 21;322(5905):1247-50.
- Role of Wnt/b-catenin signaling in drug resistance of pancreatic cancer. Curr Pharm Des. 2012;18(17):2464-2471.
- Regulation of lung cancer cell growth and invasiveness by beta-TRCP. Mol Carcinog. 2005 Jan;42(1):18-28.
- Microenvironmental interactions between endothelial and lymphoma cells: a role for the canonical WNT pathway in Hodgkin lymphoma. Leukemia. 2017 Feb;31(2):361-372.
- Wogonoside inhibits angiogenesis in breast cancer via suppressing Wnt/β-catenin pathway. Mol Carcinog. 2016 Nov;55(11):1598-1612.
- Identification of a novel inhibitor of the canonical Wnt pathway. Mol Cell Biol. 2011 Jul;31(14):3038-3051.
- Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009 Jul;8(7):547-566.
- CCNA1 (cyclin A1). Atlas Genet Cytogenet Oncol Haematol. 2012 Nov.
- The S100A6 calcium-binding protein regulates endothelial cell-cycle progression and senescence. FEBS J. 2012 Dec;279(24):4576-88.
- Identification of cyclins A1, E1 and vimentin as downstream targets of heme oxygenase-1 in vascular endothelial growth factor-mediated angiogenesis. Sci Rep. 2016 Jul 8;6:29417-.
- A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin Cancer Res 2007. 2007;13(24):7296-7304.
- Aberrant promoter methylation of multiple genes during pathogenesis of bladder cancer. Cancer Epidemiol Biomarkers Prev. 2008;17(10):2786-2794.
- Comparison of promoter hypermethylation pattern in salivary rinses collected with and without an exfoliating brush from patients with HNSCC. PLoS One. 2012.
- A subset of Suz12/PRC2 target genes is activated during HBV replication and liver carcinogenesis associated with hepatitis B virus X protein. Hepatology. 2012;56(4):1240-1251.
- Cyclin I correlates with VEGFR-2 and cell proliferation in human epithelial ovarian cancer. Gyneco Oncol. 2012 Oct;127(1):217-222.
- Multiple cellular mechanisms related to cyclin A1 in prostate cancer invasion and metastasis. J Natl Cancer Inst. 2008 Jul 16;100(14):1022-1036.
- Cyclin A1 1197 modulates the expression of vascular endothelial growth factor and promotes hormone-dependent growth and angiogenesis of breast cancer. PLoS One. 2013;8(8):e72210-.
- Expression of follicle-stimulating hormone receptor in tumor blood vessels. N Engl J Med. 2010 Oct;363:1621-1630.
- Immunohistochemical detection of FSH receptors in pituitary adenomas and adrenal tumors. Folia Histochem Cytobiol. 2012 Oct;50(3):325-330.
- Expression of follicle-stimulating hormone receptor by the vascular endothelium in tumor metastases. BMC Cancer. 2013 May 20;13:246-.
- A novel role for FSH receptor as a tumor endothelial cell marker. Acta Endocrinologica (Buc). 2010;6(4):507-512.
- FSH receptor (FSHR) expression in human extragonadal reproductive tissues and the developing placenta, and the impact of its deletion on pregnancy in mice. Biol Reprod. 2014 Sep;91(3):74-.
- Role of the phosphatidylinositol-3-kinase and extracellular regulated kinase pathways in the induction of hypoxia-inducible factor (HIF)-1 activity and the HIF-1 target vascular endothelial growth factor in ovarian granulosa cells in response to follicle-stimulating hormone. Endocrinology. 2009 Feb;150(2):915-28.
- HIF1 activity in granulosa cells is required for FSH-regulated Vegfa expression and follicle survival in mice. Biol Reprod. 2014 Jun;90(6):135-.
- The Increased Expression of Connexin and VEGF in Mouse Ovarian Tissue Vitrification by Follicle Stimulating Hormone. Biomed Res Int. 2015;2015:397264-.
- Characterization of annexin A2 in chicken follicle development: Evidence for its involvement in angiogenesis. Anim Reprod Sci. 2015 Oct;161:104-11.
- Reactive oxygen species regulate FSH-induced expression of vascular endothelial growth factor via Nrf2 and HIF1α signaling in human epithelial ovarian cancer. Oncol Rep. 2013 Apr;29(4):1429-34.
- Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Mol Biol Cell. 2012 Jul;23(13):2412-2423.
- Attenuation of cardiac dysfunction and remodeling of myocardial infarction by microRNA-130a are mediated by suppression of PTEN and activation of PI3K dependent signaling. J Mol Cell Cardiol. 2015 Oct 13.S0022-2828(15)30082-1
- PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res. 2009;102:19-65.
- MiR-19a regulates PTEN expression to mediate glycogen synthesis in hepatocytes. Sci Rep. 2015 Jun 26;5:11602-.
- PTEN expression in endothelial cells is down-regulated by uPAR to promote angiogenesis. Thromb Haemost. 2015 Aug;114(2):379-89.
- KLF5 inhibits angiogenesis in PTEN-deficient prostate cancer by attenuating AKT activation and subsequent HIF1α accumulation. Mol Cancer. 2015 Apr 21;14:91-.
- PI3K/AKT/PTEN signaling as a molecular target in Leukemia angiogenesis. Adv Hematol. 2012;2012:843085-.
- PI3K/Akt signaling in osteosarcoma. Clin Chim Acta. 2015 Apr 15;444:182-92.
- Association of high microvessel αvβ3 and low PTEN with poor outcome in stage 3 neuroblastoma: rationale for using first in class dual PI3K/BRD4 inhibitor, SF1126. Oncotarget. 2016 Nov 18;8(32):52193-52210.
- PI3K/AKT/mTOR pathway in angiogenesis. Front Mol Neurosci. 2011 Dec;4:51-.
- The PI3K/AKT/mTOR pathway is activated in gastric cancer with potential prognostic and predictive significance. Virchows Arch. 2014 Jul;465(1):25-33.
- Bmi-1 promotes invasion and metastasis, and its elevated expression is correlated with an advanced stage of breast cancer. Mol Cancer. 2011 Jan28;10(1):10-.
- Expression level of Bmi-1 oncoprotein is associated with progression and prognosis in colon cancer. J Cancer Res Clin Oncol. 2010 Jul;136:997-1006.
- Correlation of Bmi-1 expression and telomerase activity in human ovarian cancer. Br J Biomed Sci. 2008;65:172-177.
- The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009 Dec;119(12):3626-3636.
- Oncoprotein Bmi-1 renders apoptotic resistance to glioma cells through activation of the IKK-nuclear factor-kappa B Pathway. Am J Pathol. 2010 Feb;176(2):699-709.
- Bmi-1 promotes glioma angiogenesis by activating NF-kB signaling. PLoS One. 2013;8(1):e55527-.
- Bmi-1 promotes the aggressiveness of glioma via activating the NF-kappaB/MMP-9 signaling pathway. BMC Cancer. 2012 Sep 11;12:406-.
- Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. 2014 Feb;16(2):137-46.
- Interfering with Gal-1-mediated angiogenesis contributes to the pathogenesis of preeclampsia. Proc Natl Acad Sci U S A. 2013 Jul 9;110(28):11451-6.
- Immunohistochemical expression of galectin-3 in cancer: a review of the literature. Turk Patoloji Derg. 2012;28(1):1-10.
- Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009 Sep 8;1288:116-124.
- Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J Exp Med. 2010 Aug 30;207(9):1981-1993.
- Galectin-3 acts as an angiogenic switch to induce tumor angiogenesis via Jagged-1/Notch activation. Oncotarget. 2017 Jul 25;8(30):49484-49501.
- Control of angiogenesis by galectins involves the release of platelet-derived proangiogenic factors. PLoS One. 2014 Apr 30;9(4):e96402-.
- Galectin-3 accelerates M2 macrophage infiltration and angiogenesis in tumors. Am J Pathol. ;182(5):1821-1831.
- Galectin-3 disruption impaired tumoral angiogenesis by reducing VEGF secretion from TGFβ1-induced macrophages. Cancer Med. 2014 Apr;3(2):201-14.
- Cleavage of galectin-3 by matrix metalloproteases induces angiogenesis in breast cancer. Int J Cancer. 2010 Dec;127(11):2530-2541.
- Regulation of prostate cancer progression by galectin-3. Am J Pathol. 2009 Apr;174(4):1515-1523.
- The expressions and clinical significances of tissue and serum galectin-3 in pancreatic carcinoma. J Cancer Res Clin Oncol. 2012 Jun;138(6):1035-43.
- Galectin-3 contributes to melanoma growth and metastasis via regulation of NFAT1 and autotaxin. Cancer Res. 2012 Nov 15;72(22):5757-66.
- Role of pro-angiogenic marker galectin-3 in follicular neoplasms of thyroid. Indian J Biochem Biophys. 2012 Oct;49(5):392-394.
- A galectin-3-dependent pathway upregulates interleukin-6 in the microenvironment of human neuroblastoma. Cancer Res. 2012 May 1;72(9):2228-2238.
- Galectin-1 and galectin-3: plausible tumour markers for oral squamous cell carcinoma and suitable targets for screening high-risk population. Clin Chim Acta. 2015 Mar 10;442:13-21.
- Silencing of galectin-3 represses osteosarcoma cell migration and invasion through inhibition of FAK/Src/Lyn activation and β-catenin expression and increases susceptibility to chemotherapeutic agents. Int J Oncol. 2015 Jan;46(1):185-94.
- Cell surface interaction of annexin A2 and galectin-3 modulates epidermal growth factor receptor signaling in Her-2 negative breast cancer cells. Mol Cell Biochem. 2016 Jan;411(1-2):221-33.
- Senescent mesenchymal stem cells promote colorectal cancer cells growth via galectin-3 expression. Cell Biosci. 2015 May 6;5:21-.
- Hypoxia Up-Regulates Galectin-3 in Mammary Tumor Progression and Metastasis. PLoS One. 2015 Jul 29;10(7):e0134458-.
- Advanced glycation end products induce choroidal endothelial cell proliferation, matrix metalloproteinase-2 and VEGF upregulation in vitro. Graefes Arch Clin Exp Ophthalmol. 2002 Dec;240(12):996-1002.
- Advanced glycation end products (AGEs) and their receptors (RAGEs) in diabetic vascular disease. Medicographia. 2009;31:257-265.
- Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci. 2011 Dec;1243:88-102.
- Suppression of receptor for advanced glycation End products improves angiogenic responses to ischemia in diabetic mouse hindlimb ischemia model. ISRN Vascular Medicine. 2013;2013(7):908108-.
- Advanced glycation end-products induced VEGF production and inflammatory responses in human synoviocytes via RAGE-NF-κB pathway activation. J Orthop Res. 2015 Oct.
- Receptors for advanced glycation end products (RAGE) is associated with microvessel density and is a prognostic biomarker for clear cell renal cell carcinoma. Biomed Pharmacother. 2015 Jul;73:147-53.
- High-mobility group box 1 and cancer. Biochim Biophys Acta. 2010 Jan-Feb;1799(1-2):131-140.
- HMGB1: roles in base excision repair and related function. Biochim Biophys Acta. 2010 Jan-Feb;1799(1-2):119-30.
- Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005 Apr;166(4):1259-63.
- Leukocyte HMGB1 is required for vessel remodeling in regenerating muscles. J Immunol. 2014 Jun 1;192(11):5257-64.
- Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene. 2013 Jan 17;32(3):363-74.
- HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumour Biol. 2015 Oct.
- HMGB1 may act via RAGE to promote angiogenesis in the later phase after intracerebral haemorrhage. Neuroscience. 2015 Jun 4;295:39-47.
- HMGB1 induces angiogenesis in rheumatoid arthritis via HIF-1α activation. Eur J Immunol. 2015 Apr;45(4):1216-27.
- HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol. 2010;28:367-388.
- HMGB1: a two-headed signal regulating tumor progression and immunity. Curr Opin Immunol. 2008 Oct;20(5):518-23.
- Inflammation and cancer: a double-edged sword. Cancer Cell. 2007 Oct;12:300-301.
- Prognostic value of HMGB1 overexpression in resectable gastric adenocarcinomas. World J Surg Oncol. 2010 Jun 26;8:52-.
- High mobility group box 1 (HMGB1) acts as an "alarmin" to promote acute myeloid leukaemia progression. Oncoimmunology. 2018 Feb 27;7(6):e1438109-.
- Differential effects of leptin and adiponectin in endothelial angiogenesis. J Diabetes Res. 2015;2015:648239-.
- Leptin and its cardiovascular effects: Focus on angiogenesis. Adv Biomed Res. 2015 May;4:79-.
- Leptin-cytokine crosstalk in breast cancer. Mol Cell Endocrinol. 2014 Jan 25;382(1):570-582.
- Leptin is a physiological regulator of skeletal muscle angiogenesis and is locally produced by PDGFRα and PDGFRβ expressing perivascular cells. Angiogenesis. .2018Aug18
- Hypoplastic left heart syndrome is associated with structural and vascular placental abnormalities and leptin dysregulation. Placenta. 2015 Oct;36(10):1078-86.
- Leptin promotes wound healing in the skin. PLoS One. 2015 Mar 23;10(3):e0121242-.
- Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim Biophys Acta. 2012 April;1825(2):207-222.
- Obesity and cancer risk: evidence, mechanisms, and recommendations. Ann NY Acad Sci. 2012 Oct;1271:37-43.
- Leptin attenuates the anti-estrogen effect of tamoxifen in breast cancer. Biomed Pharmacother. 2013 feb;67(1):22-30.
- Involvement of leptin receptors expression in proliferation and neoangiogenesis in colorectal carcinoma. J BUON. 2015 Jan-Feb;20(1):100-8.
- Leptin acts on neoplastic behavior and expression levels of genes related to hypoxia, angiogenesis, and invasiveness in oral squamous cell carcinoma. Tumour Biol. 2017 May;39(5):1010428317699130-.
- Integrative proteomics and tissue microarray profiling indicate the association between overexpressed serum proteins and non-small cell lung cancer. PLoS One. 2012;7(12):e51748-.
- Proteomic identification of exosomal LRG1: a potential urinary biomarker for detecting NSCLC. Electrophoresis. 2011 Aug;32(15):1976-1983.
- Serum leucine-rich alpha-2 glycoprotein is a disease activity biomarker in ulcerative colitis. Inflamm Bowel Dis. 2012 Nov;18(11):2169-2179.
- The role of TGFβ1 and LRG1 in cardiac remodelling and heart failure. Biophys Rev. 2015;7:91-104.
- LRG1 promotes angiogenesis by modulating endothelial TGF-b signaling. Nature. 2013;499(7458):306-311.
- Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS One. 2011 Feb 7;6(2):e14665-.
- Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc Natl Acad Sci USA. 2002 Jan 22;99(2):715-20.
- Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res. 2012 Apr 27;110(9):1217-1225.
- Direct Activation of NADPH Oxidase 2 by2-Deoxyribose-1-Phosphate Triggers Nuclear Factor Kappa B-Dependent Angiogenesis. Antioxid Redox Signal. 2018 Jan 10;28(2):110-130.
- NADPH oxidase 1 plays a critical mediating role in oncogenic Ras-induced vascular endothelial growth factor expression. Oncogene. 2008 Aug 7;27(34):4724-32.
- Reactive oxygen generated by NADPH oxidase 1 (Nox1) contributes to cell invasion by regulating matrix metalloprotease-9 production and cell migration. J Biol Chem. 2010 Feb 12;285(7):4481-8.
- Role of urotensin II in health and disease. Am J Physiol Regul Integr Comp Physiol. 2010 May;298(5):R1156-R1172.
- Human urotensin II promotes hypertension and atherosclerotic cardiovascular diseases. Curr Med Chem. 2009;216:550-563.
- Urotensin II induces interleukin 8 expression in human umbilical vein endothelial cells. PLoS One. 2014 Feb 21;9(2):e90278-.
- The HIF1 target gene NOX2 promotes angiogenesis through urotensin-II. J Cell Sci. 2012;125:956-964.
- Signaling switch of the urotensin II vasoactive peptide GPCR: prototypic chemotaxic mechanism in glioma. Oncogene. 2015 Sep 24;34(39):5080-94.
- The role of the UTS2 gene polymorphisms and plasma Urotensin-II levels in breast cancer. Tumour Biol. 2015 Jun;36(6):4427-32.
- STIMulating store-operated Ca2+ entry. Nat Cell Biol. 2009 Jun;11(6):669-677.
- STIM1 gates the store-operated calcium channel orai1 in vitro. Nat Struct Mol Biol. 2010 Jan;17(1):112-6.
- Increased activation of stromal interaction molecule-1/Orai-1 in aorta from hypertensive rats: a novel insight into vascular dysfunction. Hypertension. 2009 Feb;53(2):409-16.
- Expression of Orai genes and I(CRAC) activation in the human retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 2011 Jan;249(1):47-54.
- Molecular and functional characterization of ORAI and STIM in human corporeal smooth muscle cells and effects of the transfer of their dominant-negative mutant genes into diabetic rats. J Urol. 2012 May;187(5):1903-10.
- Orais and STIMs: physiological mechanisms and disease. J CellMol Med. 2012 Mar;16(3):407-424.
- Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011 May 13;108(10):1190-1198.
- Orai1 and transient receptor 1011 potential channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma 1012 and other malignancies. Anticancer Agents Med Chem. 2014 Feb;14(2):296-312.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.