The SARS-CoV2 - ACE2 link: a physiopathological analysis
Vascular Cell. 2020;
Received: 18 June 2020 | Accepted: 22 July 2020 | Published: 03 August 2020
Vascular Cell ISSN: 2045-824X
Abstract
One Sentence Summary
The interaction between SARS-CoV2 and its functional receptor ACE2 determines a variety of severe effects such as systemic inflammation, endothelial dysfunction, microangiopathy, myocarditis, thrombosis and myocardial infarction which define a predominant cardiovascular form of Covid-19.
Abstract
The Covid-19 pandemic represents an unsolved problem which has caused numerous fatalities. At the present time, there is no vaccination available or curative therapy. However, recent reports suggest that SARS-CoV2 acts upon its functional receptor ACE2 inducing a variety of deleterious effects such as inflammation, endothelial dysfunction, microangiopathy, myocarditis, thrombosis and myocardial infarction. The details of the SARS-CoV2 -ACE2 interaction are poorly understood and most of the hypothesis related to this are based on the extrapolation of previous research focusing on ACE2 as a receptor for SARS-CoV. Considering the similarities between SARS-CoV2 and SARS-CoV we have conducted a physiopathological analysis focusing on the key pathogenic role of ACE2 as a functional receptor for SARS-CoV2. In this context, we have identified several potential therapeutic targets which should be further evaluated in patients with Covid-19. It is likely that an efficient therapy for Covid-19 will be revealed by research investigating the binding of viral spike S protein to ACE2, and the immunological response determined by SARS-CoV2-ACE2 interaction, including the anti-viral role of plasmacytoid dendritic cells and anti-inflammatory reprogramming of macrophages.
Keywords
Introduction
The COVID-19 pandemic originating in Wuhan, China represents the most important public health issue of this century, threatening the national security and integrity of numerous countries around the world. In the past, the medical world was cautioned about the possibility of serious coronavirus outbreaks due to the high ability of these viruses for recombination. In addition, these viruses are extensively used in research and they are seen commonly in bats and other exotic animals which are consumed traditionally in China [1].
In general, coronaviruses have an extremely high degree of infectivity, the 2002-2003 SARS outbreak been tracked down to a single individual working in the medical field in Guangdong, China which was infected with the Urbani strain of coronavirus (SARS coronavirus/SARS-CoV). Additional research has indicated that SARS-CoV represents the result of recombination of two different bat viruses and exerts its pathogenic effect after interaction with angiotensin converting enzyme 2 (ACE2) receptors, a homologue of angiotensin converting-enzyme (ACE) [2, 3, 4]. However, compared with ACE which controls the synthesis of Angiotensin II (Ang II) from Angiotensin I (Ang I), ACE2 acts as a negative modulator of the renin-angiotensin system promoting transformation of Ang II into Angiotensin 1-7 (Ang 1-7) [4, 5]. Remarkably, the high infectivity of SARS-CoV could be explained by the structure of its spike S protein. This shows a unique receptor binding motif which was acquired by recombination followed by mutation, providing a strong and efficient SARS-CoV-ACE2 interaction [3].
In the context of the current Covid-19 pandemic, post-mortem clinical studies suggest that SARS-CoV2 may also act upon the ACE2 inducing a variety of clinical symptoms affecting not only the respiratory function but also the cardiovascular and immunological systems [6]. Considering the similarity between SARS-CoV and SARS-CoV2, we will review the current relevant data analyzing the physiopathological interaction between the SARS-CoV2 and the ACE2, describing the most important factors which may have a potential therapeutic role.
The renin-angiotensin system (RAS)
The renin-angiotensin system (RAS) has a role of paramount importance maintaining normal blood pressure. It is already well known that in in patients with heart failure, renin induces an increased production of angiotensin I (Ang I) from angiotensinogen. Angiotensin converting enzyme (ACE) transforms angiotensin I (Ang I) peptide in angiotensin II (Ang II) which has a significant role in hypertension, atherosclerosis and heart failure. The Ang II induces its pathological vasoconstrictive, pro-inflammatory and pro-fibrotic effects via G-protein factors such as the angiotensin II type 1 receptors (AT1R) and angiotensin II type 2 receptors (AT2R) [7]. Previous studies conducted in vitro and in vivo have revealed that vascular overload results in an increased release of Ang II followed by activation of AT1R which induces apoptosis in cardiac cells exposed to increased vascular volumes while the ATR2 is also over expressed after myocardial ischemia [8, 9, 10].
Also, Ang II is active via non-G pathways, such as Janus kinases, focal adhesion kinase (FAK) and c-Src as well as via platelet-derived growth factor (PDGF), epidermal growth factor receptor (EGFR), and insulin growth factor receptor (IGFR) modulating a variety of biological effects including vascular inflammation and production of reactive oxygen species [11]. Experimental studies evaluating phosphatidylinositol 3-kinase (PI3-kinase), have reported a rapid phosphorylation of the insulin growth factor-I (IGF-1) receptor after administration of Ang II suggesting a direct action resulting in insulin resistance. However, this effect was blocked by losartan, an AT1R blocker which also stops phosphorylation of Src kinase, subsequently improving insulin sensitivity [12]. This is of paramount importance as insulin resistance is associated with an increased risk of cardiovascular disease. However, Ang II is responsible for activation of other non-G pathways such as mitogen-activated protein kinase (MAPK), Raf, paxillin, Shc, Akt/protein kinase B and phospholipase C-gamma, inhibiting the vasorelaxant NO while in the same time increasing the level of intracellular reactive oxygen species (ROS) [13].
It is very important to highlight that Ang II acts also upon G -protein AT1R and AT2R promoting ROS synthesis. Moreover, ROS act in concert with Ang II activating MAP kinases, protein tyrosine phosphatases, tyrosine kinase as well as RhoA/Rho cellular kinases enhancing vascular contraction, vascular senescence and inflammation [14]. Additional damage may also be induced by Ang II activation of Map kinases/ATF-2 which promote upregulation of endothelial arginase and decrease of endothelial NO resulting in deficient vasodilation [15] (Figure 1).
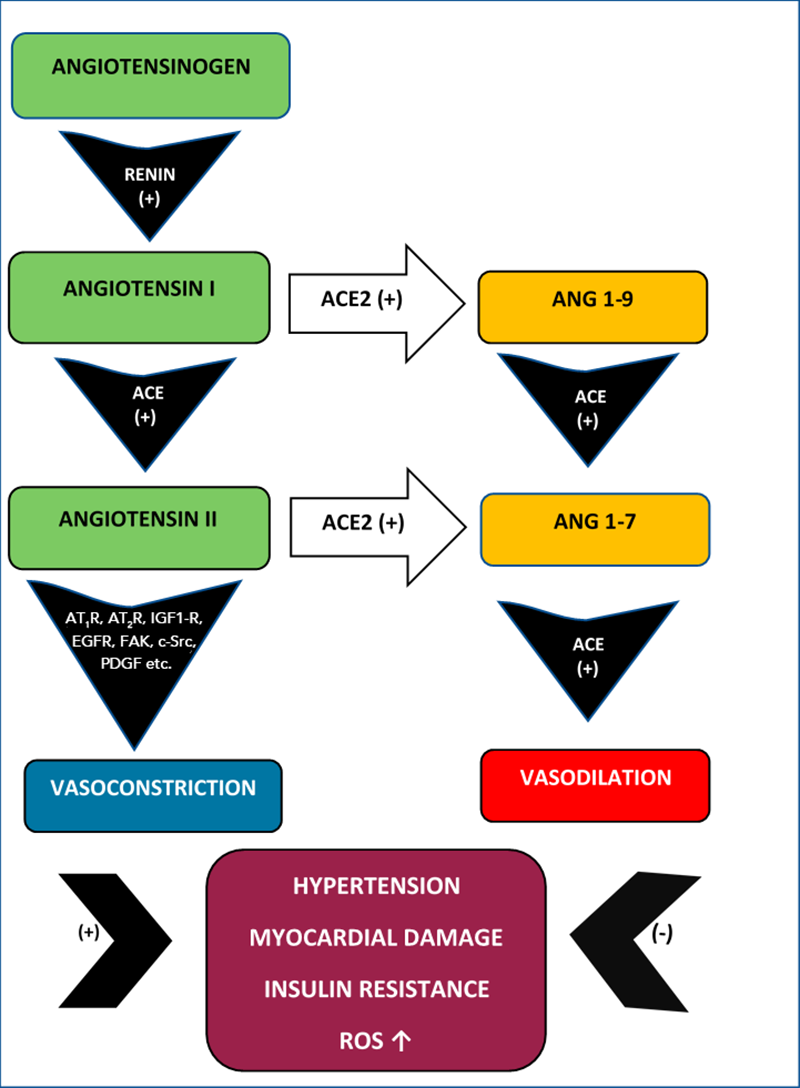
Figure 1
Figure 1
Ang I-Ang II vs Ang 1-9-Ang 1-7. The vasoconstrictor Ang II promotes hypertension, myocardial damage, insulin resistance and ROS upregulation while the Ang 1-7 has a vasodilator action counteracting these effects.
ACE2 – Ang 1-7 - Mas system
Recent research suggests that RAS is closely associated with ACE2, a type I cellular membrane protein (carboxypeptidase) which displays a 40% identity and 60% similarity with ACE. Also, it is known that ACE2 displays an active binding site on the extracellular endothelial surface [16]. Apart from lung, the largest concentration of ACE2 is identified in the enterocytes of the small intestine. Also, ACE2 is expressed on vascular endothelial cells, smooth muscle cells as well as in a variety of other tissues and organs such as: oral and nasal mucosa, nasopharynx, thymus, skin, heart, kidney, brain, testis, spleen, bone marrow and lymph nodes, monocytes and macrophages [2, 17, 18]. Anyway, most of the ACE2 is located on the cellular membrane and a very small amount is circulating as a soluble form [19, 20, 21].
It seems that ACE2 is overexpressed in various cardiovascular conditions such as heart failure. Donoghue et al (2000) have indicated that ACE2 is increased in human myocardium with morphological changes characteristic of ventricular failure as well as on the coronary and renal vascular endothelial cells. This specific localization indicates that ACE2 could have potentially a significant role maintaining the cardiovascular haemodynamic functions [22]. Moreover, it was reported that a deficiency of ACE2 is associated with abnormal vascular contractility [9, 13].
Initially, clinical studies conducted in patients which received a donor heart, showed that intracoronary administration of Ang I, increases the levels of angiotensin 1-7 (Ang 1-7) which has vascular relaxant action. In the meantime, the vasodilator Ang 1-7 synthesis decreases significantly after suppression of vasoconstrictor Ang II production [23]. However, these findings can be explained by the modulating effects of ACE2. More specifically, in the context of RAS, ACE2 acts as a natural modulator of the vasoconstrictor Ang II by promoting its transformation into the vasodilator Ang 1-7 [18]. However, Ang I is metabolized into another precursor, the angiotensin 1-9 (Ang 1-9) in the presence of ACE2 [20, 24, 25, 26] (Figure 1)
However, the balance between the vasodilator system ACE2-Ang 1-7 and vasoconstrictor Ang II is provided by the aldosterone. Studies conducted in cultured rat myocardial cells have indicated that ACE2 level is decreased by aldosterone which in parallel, upregulates the ACE and Ang II synthesis [27]. Interestingly, ACE2 synthesis is not blocked by captopril or lisinopril, which are already used in clinical practice as anti-hypertensive agents with ACE inhibitory action [16, 17].
However, subsequent studies conducted both in animal models and patients with cirrhosis have confirmed that ACE2 induced increase in Ang 1-7 determines activation of Mas receptor which results in splanchnic vasodilation [28]. Also, activation of Mas receptor prevents vasoconstriction in the heart, kidney and brain [26, 29]. Other studies have confirmed that Mas receptors control the vascular tone of the vessels promoting vasorelaxation potentially having a significant role in the pathogenesis of atherosclerosis and hypertension [30]. Moreover, previous experimental studies conducted in a Mas deficient mouse model have indicated that in these subjects the Ang 1-7 cannot exert its vasorelaxant and anti-diuretic effects, indicating that in fact Mas represents a receptor for Ang 1-7 [31]. Therefore, Ang 1-7 should be considered as an endogenous agonist for Mas receptors activating a chain of vasorelaxant effects. However, the exact mechanism of inter-play between various factors involved in ACE2-Ang 1-7-Mas pathway are not completely understood and more research is currently required [32].
Experimental research conducted in transgenic mice which are characterised by an increased level of ACE2 synthesis has revealed that those subjects who have developed ischemic stroke after oxygen and glucose deprivation but were treated with Ang 1-7 had a much better outcome than those that were not treated with this Mas receptor agonist. The authors recorded in treated subjects significant less neuronal death and decreased formation of reactive oxygen species (ROS) and NADPH oxidases. Cerebral oedema was less obvious in subjects treated with Ang 1-7. Overall, this data would suggest that the clinical features and the rate of recovery in those with ischemic stroke patients depend also on the ability of the ACE2/Ang 1-7/Mas vasorelaxant pathway to counteract the vasoconstrictive Ang I -ACE- Ang II-AT1R system [33].
ACE2 as a SARS-CoV receptor
In general, viral cellular infectivity depends on the presence of cellular surface proteins which can act as functional receptors. Initial experiments conducted on coronaviruses, have indicated that ACE2 has been isolated from cells infected with SARS-CoV. In this context, SARS-CoV binds ACE2 through its viral spike S protein. Also, in vitro studies have confirmed that SARS-CoV can replicate in cells previously transfected with ACE2. However, SARS-CoV replication is blocked by ACE2 antibodies suggesting that ACE2 is a functional receptor for this SARS-CoV [34]. Subsequent studies have indicated that ACE2 has a physiological role as a receptor not only for SARS-CoV but also for other coronaviruses such as NL63 and the newly identified SARS-CoV2 [19, 20, 21].
Most importantly, respiratory mucosa represents the best environment for SARS-CoV development who binds the ACE2 on the apical surface of epithelial respiratory cells especially on type II pneumocytes [35]. In addition, it has been established that in human lung parenchyma, SARS-CoV-S protein promotes ACE2 shedding and lung parenchyma destruction [36, 37].On another perspective, research conducted in ACE2 knockout mice has indicated that in a murine model of ARDS, ACE2 has a local protective role preventing lung parenchyma damage while administration of an extract of SARS-CoV spike S protein promotes lung destruction. However, inhibition of RAS and Ang II significantly decreases respiratory failure [4].
SARS-CoV2: physiopathological pathways
It appears that the most important aspect of SARS-CoV2- ACE2 interaction is represented by the inhibition of the ACE2-Ang 1-7-Mas vasodilator pathway. As a result, the unopposed action of Ang II could potentially result in vasoconstriction, inflammation and immunological abnormalities, endothelial dysfunction-microangiopathy, myocarditis, thrombosis and myocardial infarction. At the present time, a significant number of studies support a causal relationship between SARS-CoV2-ACE2 and the above-mentioned clinical effects. However, most of these studies are limited due to small number of subjects evaluated or their findings are yet to be verified in animal models or patients with Covid-19. An assessment from a physiopathological perspective, of the most promising of these studies suggesting either a potential therapeutic target or a category of patents at increased risk of developing severe Covid-19 follows below.
a. ACE2-SARS-CoV2 interaction
The SARS-CoV2 has a 30 kb single-stranded positive-sense RNA, similar with the coronavirus from pangolin, horseshoe-bat as well as with the previously described SARS-CoV. At the present time, it is believed that SARS-CoV2 has a significant ability for recombination and inter-species transmission [38]. Although, the etiopathogenic mechanism of SARS-CoV2 is not fully understood, its interaction with ACE2 determines a wide-range of effects of significant importance for clinical evolution of Covid-19 patients [39].
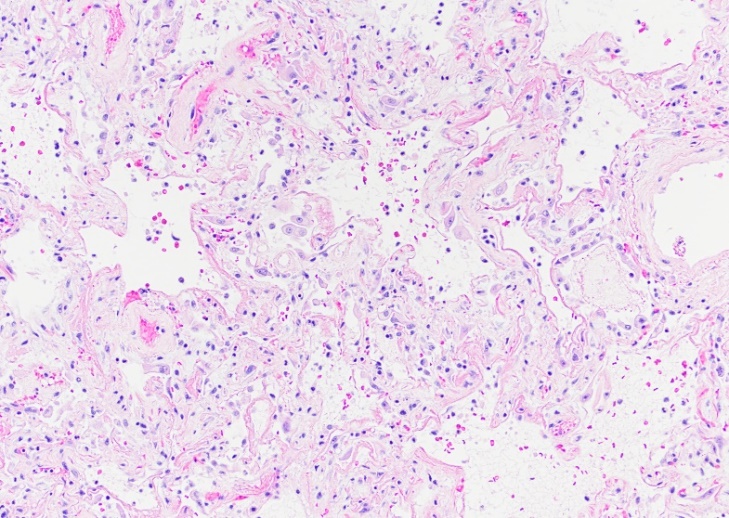
Therefore, the symptomatology in Covid-19 varies from a simple cold and flu-like features to atypical pneumonia with high fever and in some cases severe hypoxemia, respiratory failure and death [40]. The severe acute respiratory distress syndrome produced by SARS-CoV2 translates histologically into diffuse alveolar damage characterized by edematous widening of the alveolar septa associated with alveolar collapse, inflammation and deposition of hyaline membranes (Figure 2). However, these morphological changes are not pathognomonic for SARS-CoV2 infection.
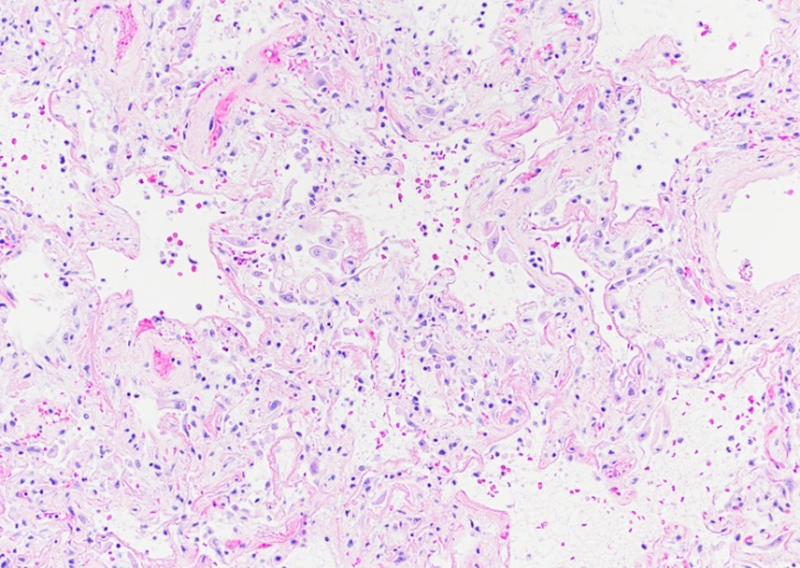
Figure 2
Figure 2
SARS-CoV2 diffuse alveolar damage (DAD). Widening of the alveolar septa, inflammation and proteinaceous material deposition.
Recent reports suggest that Covid-19 is associated with severe cardiovascular complications such as: endothelial dysfunction, microangiopathy, myocarditis, thrombosis and disseminated intravascular coagulation, heart failure and pulmonary embolism [41].
Extrapolating the information revealed by previous studies evaluating the SARS-CoV action on ACE2 receptors, we could hypothesize that cardiovascular complications in Covid-19 patients represent the result of the SARS-CoV2-ACE2 interaction. However, this is not entirely satisfactory as SARS-CoV and SARS-CoV2 differ in the structure of their ACE2 receptor binding domain (RBD) of the S protein. In the case of SARS-CoV2, its RBD is compact stabilizing two binding spots located at the junction of S protein with ACE2 protein. It seems that this particularity offers SARS-CoV2 an increased ACE2 binding affinity compared with SARS-CoV [42].
Recently, Coutard B et al (2020) have indicated that the newly described SARS-CoV2, presents with another structural particularity which may explain its pathogenicity. The authors have identified a furin-like cleavage located at the level of S-protein which could facilitate infectivity. This special morphology is also seen present in MERS-CoV-S protein. Interestingly, it seems that other viruses such as HIV use furin for cellular invasion. Therefore, it was proposed that the furin-like cleavage site supports the S-protein priming functioning as a switch-on mechanism for SARS-CoV2 infectivity and intrusion into the cells [43]. In addition, the internalization of SARS-CoV2 is facilitated by transmembrane protease serine 2 (TMPRSS2) which is used by the virus for the priming of the spike S protein. However, the SARS-CoV2 cellular penetration can be efficiently blocked by a clinically tested pharmacological agent, more specifically an inhibitor of TMPRSS2 such as camostate mesylate which is already approved for clinical pancreatitis and drug induced injury in the lung [44].
Moreover, recent reports suggest that a specific mutation of the viral spike S-protein is responsible for increased infectivity observed in Covid-19 patients. In vitro studies using HEK293T cells modified to display human ACE2, have indicated that SARS CoV2 samples with a D614G mutation in the S-protein, have an increased rate of infectivity of these cells. This was associated with increased rates of inclusion of the viral protein into the pseudo-virion. Overall it seems that the SARS-CoV2 strains with D614G are spread with greater efficiency and show an increased level of stability [45]
Recently, some authors have indicated that in hypertension as well as smoking and diabetes, which appear to be associated with a severe form of Covid-19 disease, the ACE2 gene is upregulated [46, 47]. A recent study reports that both controlled allergic exposure as well as asthma are associated with a low ACE2 level, suggesting that this group of patients would develop mild forms of disease. It is believed that interleukin 13 (IL-13) which is released in patients suffering from allergic asthma, significantly decreases ACE2 level in the bronchial and nasal mucosa [47]. However, while the relationship between IL-13 and ACE2 was tested in adults and children with allergic asthma, this hypothesis has not been tested in those with concomitant allergic asthma and Covid-19. Therefore, it should be regarded with great caution.
Also, animal studies have shown that ACE inhibitors and Angiotensin II Receptor Blockers increase the level of ACE2 synthesis. Based on this information, it was hypothesised that an increased number of SARS-CoV2 particles could bind the available ACE2 functional receptors [48, 49]. Other studies conducted in a murine model of SARS-CoV induced pneumonia have shown that Angiotensin II Receptor Blockers may prevent the development of lung damage [48]. In this context, Verdecchia P et al (2020) indicates that although, the treatment with ACE inhibitors and Angiotensin II Receptor Blockers is followed by increased Ang 1-7 and ACE2 mRNA, there is no clinical evidence to suggest that an increased ACE2 would represent a risk for acquiring SARS-CoV2 infection by providing more binding sites for the virus [21]. Moreover, other groups have reported no ACE2 upregulation in patients treated with ACE inhibitors and Angiotensin II Receptor Blockers [50]. However, due to the limited number of enrolled subjects, the resuts f these studies must be regarded with caution.
Interestingly, experimental studies conducted in animal models have revealed that SARS-CoV2 downregulates the ACE2 functional receptors but only in the lung, not in other tissues [21]. Subsequently the whole ACE2-Ang 1-7-Mas pathway is inhibited, ultimately resulting in a decrease of the endogenous vascular relaxant and anti-inflammatory effects of Ang 1-7 while the vasoconstrictive Ang II pathway remains fully functional, promoting inflammation, thrombosis and pulmonary oedema with respiratory insufficiency. As expected, in experimental studies, these Ang II mediated effects seem to be prevented by administration of Angiotensin II Receptor Blockers [51].
b. Inflammation associated with SARS-CoV2
It has been reported that ACE2-Ang 1-7-Mas system decreases the levels of numerous pro-inflammatory cytokines preventing lung inflammation and oedema, decreasing endothelial cell activation and pneumocyte apoptosis [52]. Moreover, by acting on Mas receptors located on macrophages, Ang 1-7 induces an anti-inflammatory reprogramming of these cells [53]. Therefore, as Covid-19 is associated with a blockage of ACE2-Ang 1-7-Mas we could speculate that this disease may be associated with an overexpression of pro-inflammatory cytokines.
It is possible that following infection with SARS-CoV2, the cells die and release virions and cellular components which determine an inflammatory-immunological reaction. Both SARS-CoV and SARS-CoV2, may be recognized by melanoma differentiation-associated protein 5 (MDA5), Toll-like receptors (TLR-)3 and 7 and cytoplasmic RNA sensors. MDA5 activates an intracytoplasmic factor, interferon regulatory factor 3 (IRF3) which increases production of interferon, IL1, IL6 and TNF-alpha [54, 55]. Interestingly, recent clinical observations in patients confirmed with Covid-19 indicate that an IL-6 peak is correlated with severe respiratory symptomatology [56].
Anyway during SARS-CoV viral infection, interferon regulatory factor 3 (IRF3) works in concert with interferon regulatory factor 7 (IRF7), activator protein (AP)-1, activating transcription factor (ATF)-2/jun, nuclear factor of activated T cells (NF-AT) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) inducing an inflammatory chain of reactions [57]. TLR-3 and TLR-7 activate NFkB which promote an increased synthesis of interferon, IL-1, IL-6, IL-8 and TNF-alpha. Also, during viral infections, human macrophages seem able to synthesise and release CXCL10, a very strong antiviral factor. Ultimately, all of these factors, theoretically, would allow the organism to counteract and remove the SARS-CoV and perhaps also the SARS-CoV2 [54, 55]. Therefore, evaluation of these factors in the setting of Covid-19 may the development of new improved therapeutic avenues.
Apart from fighting a viral infection, the immunological response may also be detrimental. Previous studies conducted in patients with SARS-CoV and MERS-CoV have indicated that severe forms of disease including respiratory failure is associated with over-expression of pro-inflammatory cytokines [57]. Moreover, clinical studies conducted in patients with SARS-CoV have revealed in fatal cases, a high-level of interferon stimulated CXCL10, CCL2 genes throughout the duration of disease but with a low level of anti-S protein antibodies [58]. However, we do not know if Covid-19 is also characterized by a low level of anti-S protein antibodies.
Kindler E et al (2016), indicate that initially SARS-CoV infection is associated with a very small production of interferon detectable after a significant delay. In this context, the “delay in interferon production” allows accumulation of numerous monocytes and macrophages secreting cytokines which induce vascular destruction in the lung parenchyma [59]. Considering all these and the similarity between SARS-CoV and SARS-CoV2, it is possible that the “delay in interferon production” could potentially explain the deterioration seen in some Covid-19 patients.
Moreover, in vitro experiments using protein localization and chromatin immunoprecipitation have indicated a SARS-CoV papain-like protease inhibits IRF3 phosphorylation subsequently inhibiting interferon production [60]. To date, the presence of a SARS-CoV2 papain-like protease remains to be established.
In addition, SARS-CoV blocks production of interferon via inhibition of interferon regulatory factor 3 and 7 (IRF3, IRF7) action by several of its viral proteins such as non-structural proteins (nsp1, nsp3, nsp7, nsp15), ORF3b, ORF6, and ORF9b [55, 61, 62]. Considering all these results, it would be very interesting to verify if these viral proteins are also seen in SARS-CoV2 as they could potentially represent therapeutic targets in the setting of Covid-19. However, a significant number of these experiments are limited as they have been conducted in vitro and are yet to be confirmed in animal models or patients.
Interestingly, in the context of a SARS-CoV it seems that plasmacytoid dendritic cells are still able to synthesise interferon [63, 64]. Unfortunately, at the present time, we have no information about the functionality of the plasmacytoid dendritic cells in patients with Covid-19 and more information is required urgently as manipulation of these cells could be of paramount importance in this setting.
c. Endothelial dysfunction-microangiopathy associated with SARS-CoV2.
A recent post-mortem study conducted in 21 patients diagnosed with Covid-19 has revealed diffuse alveolar damage and thrombosis. Some patients also showed evidence of bronchopneumonia, alveolar haemorrhage and vasculitis. Three patients included in this study developed a generalized form of thrombotic microangiopathy [65]. Other recent studies conducted in patients with SARS-CoV2 report that these patients develop thrombotic microangiopathy associated with intravascular coagulopathy [66].
At the present time the link between SARS-CoV2 and ACE2 and the risk for developing microangiopathy has not been evaluated in clinical or experimental studies. However, it is possible that SARS-CoV2 downregulates the ACE2-Ang 1-7-Mas pathway while the Ang II remains unopposed inducing vascular inflammation and thrombosis [21]. This could be plausible as the role of Ang II in promoting vascular angiopathy has been confirmed by previous studies conducted in patients with diabetic angiopathy and rheumatoid vasculitis which proved that microangiopathy is determined by an over-production of Ang II [67]. In addition, captopril, an ACE inhibitor which blocks transformation of Ang I in Ang II, prevents also the development of polyarteritis nodosa, a form of necrotizing vasculitis [68].
The etiopathogenic role of SARS-CoV2 promoting vascular inflammation and microangiopathy by blocking the ACE2-Ang 1-7-Mas pathway is also supported by previous studies conducted in ACE2 knock-out mice who have indicated that endothelial dysfunction in cerebral arteries is directly determined by a lack of ACE2 [69]. Interestingly, in experiments conducted in a murine model of stress, the subjects had an increased sensitivity to Ang II which was associated with a significant vascular inflammation [70]. However, in some clinical conditions such as retinal vascular disease, vascular inflammation is induced by Ang II only if associated with an overexpressed IL-6, a factor which is increased in infections with both SARS-CoV and SARS-CoV2 [55, 56].
d. Myocarditis associated with SARS-CoV2
The development of myorcaditis has already been documented in fatal cases of SARS-CoV. Oudit GY et al (2009) have reported that SARS-CoV viral RNA was present in 35% of the myocardium removed for evaluation from patients who died during the SARS epidemic in Toronto (2002-2004). The histopathological examination of the SARS-CoV viral RNA positive myocardium samples confirmed a significant level of cardiac inflammation characterized by an extensive infiltration of inflammatory cells while the myocardial ACE2 was significantly decreased [71]. Therefore, the interaction between ACE2 and SARS-CoV and perhaps SARS-CoV2 is of paramount importance in inducing myocarditis.
Previous research has indicated that ACE2 is overexpressed in myocardium and blood vessels in those with cardiac pathology such as aortic stenosis and heart failure [72]. Therefore, in this setting overexpression of ACE2 in pathological myocardium would theoretically offer more binding sites for SARS-CoV2 and would explain why those with cardiac conditions would develop aggressive forms of Covid-19. However, this remains to be verified in large clinical studies.
Some authors have indicated that the most likely mechanism for Covid-19 myocarditis is a direct viral injury associated with an abnormal hyperimmune reaction detrimental to the myocardial morphology and physiology [73]. A post-mortem evaluation of the myocardium of a 17-year-old male patient, confirmed post-mortem with Covid-19, who died two days after developing headache and vomiting revealed cardiomegaly and extensive eosinophilic infiltration [74]. This would indicate that SARS-CoV2 myocarditis presents initially as an acute form of eosinophilic myocarditis suggesting a hyperimmune reaction. Interestingly, an endomyocardial biopsy performed in a 43-year-old female, ten days from the start of clinical respiratory symptoms and one week after SARS-CoV2 laboratory confirmation, has indicated the presence of lymphocytic myocarditis [75]. All these data suggest that in some patients, SARS-CoV2 induces initially a hyperimmune reaction characterized by eosinophilia which after an interval of time is replaced by an adaptive immune response.
Anyway, the Covid-19 immunological myocarditis is associated with significantly increased levels of troponin I but without myocardial infarction. Overall it seems that up to 20% of Covid-19 patients may present with acute myocardial non-infarction injury determined by myocarditis. Although, routine evaluation of troponin I in patients with SARS-CoV2 is not recommended, this may change as more clinical information becomes available [76].
The long-term consequences on cardiac dynamics in those that survive after been diagnosed with SARS-CoV2 myocarditis are currently not known. However, in order to prevent any potential serious consequences including heart failure some authors recommend a close follow-up of those that have recovered after SARS-CoV2 myocarditis [77].
e. Thrombosis and Myocardial Infarction associated with SARS-CoV2
Increased levels of D-dimer and fibrin/fibrinogen represent the earliest laboratory anomalies detected in patients with Covid-19 [78]. Certainly, Covid-19 thrombosis and myocardial infarction could be explained by the binding of SARS-CoV2 on endothelial ACE2 receptors followed by endothelial activation/dysfunction, platelet aggregation and thrombosis/infarction. Other pro-thrombotic mechanisms such as platelet activation, stasis and inflammation may also work in concert promoting thrombosis [79].
However, it was reported that in fatal cases of SARS-CoV2 the endothelial damage is characterised by intracellular viral presence as well as thrombosis with angiopathy and intussusceptive angiogenesis [6]. Also, disseminated intra-vascular coagulation has been seen in Covid-19 fatal cases [80].
SARS-CoV2 thrombosis has been described in wide range of vascular territories in both arteries and veins. A study conducted in 150 patients (median age 63) diagnosed with Covid-19 found that sixty-four subjects of this cohort developed thrombosis and the most frequent clinical form was represented by pulmonary embolism (16.7%) [81]. A recent report describes a 65-year-old Covid-19 patient presenting with neurological signs resulting from cerebral venous thrombosis developed after a hemorrhagic infarct in the transverse and sigmoid sinuses [82]. Also, Le Berre et al (2020) presents a Covid-19 patient who developed concomitant pulmonary embolism and acute aortic thrombosis [83] while histopathological evaluation of five full-term placentas from patients diagnosed with Covid-19 showed thrombosis in the fetal vessels suggesting fetal vascular insufficiency [84].
In patients with Covid-19, type I myocardial infarction ensues after endothelial dysfunction with plaque fracture and thrombosis in the coronaries and type II infarction has been reported in patients with hypoxia determined by ARDS complicated with fever, tachycardia and increased oxygen cardiac demands [85].
However, previous experimental research indicates that an upregulated ACE2 protein limits the effects of a myocardial infarction [86]. Therefore, in Covid-19, it is likely that a downregulation of ACE2 receptors while the Ang II action is not opposed, may facilitate an acute coronary syndrome.
This is supported by previous studies which have shown that in human coronaries, Ang II promotes endothelial dysfunction and atheroma plaque rupture [87]. These findings have been confirmed by other studies reporting that Ang II induces overexpression of calpain, a cytosolic neutral cysteine protease which increases endothelial dysfunction increasing adhesion of leukocytes to vascular endothelium via an NF-kB mediated reaction [88].
In addition, experimental research conducted in a murine model has indicated that Ang II induces a dose-dependent endothelial dysfunction associated with increased levels of IL6 and local accumulation of vascular macrophages [89]. These data correlate with other studies reporting an increased level of IL6, Ang II, AT1R and macrophages in the shoulder region of the coronary atheroma plaque which is prone to rupture [87]. Ultimately, Ang II promotes further synthesis and release of inflammatory cytokines via activation of ROS, NADPH oxidase and NF-kB promoting an accelerated atheroma plaque development [90].
Conclusions
At the present time, the Covid-19 pandemic is far from finished and the significant differences in mortality and morbidity between countries and regions with similar medical and healthcare systems are yet to be understood. Detailed transdisciplinary studies are required urgently to identify the risk factors leading to a severe outcome in selected groups of patients. Therefore, identification of all intricacies at work inter-connecting ACE2-Ang 1-7-Mas, Ang II and other inflammatory and immunological factors may offer an opportunity for the development of an effective therapy for Covid-19.
It is known that the elderly and those with previous hypertension, heart failure, and diabetes develop severe forms of Covid-19. However, all these pathological conditions are characterized by a certain degree of systemic chronic inflammation and it has been hypothesized that they might be associated with an overexpression of endothelial ACE2 which is a functional receptor for SARS-CoV2 binding.
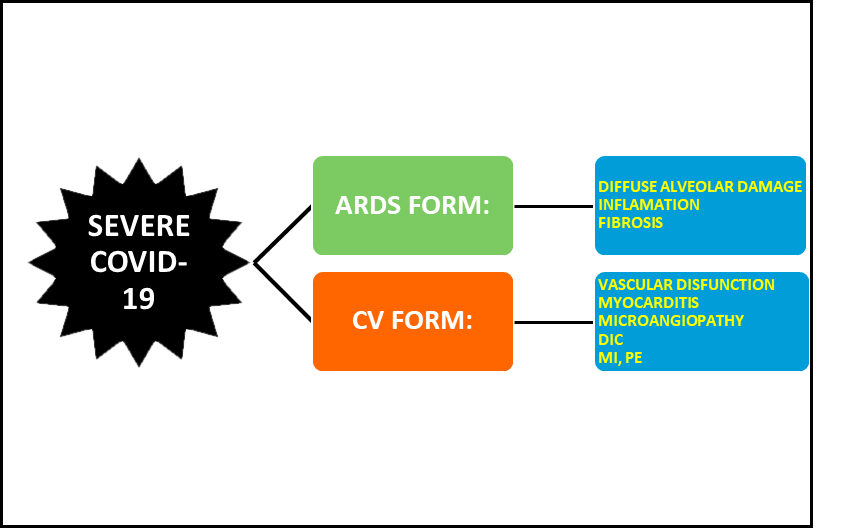
In Covid-19 patients, ACE2 downregulation by SARS-CoV2 leaves the RAS-Ang II system unopposed with numerous deleterious effects, especially in those patients with cardiovascular co-morbidities. Therefore, the patients with severe Covid-19 could either develop a severe respiratory distress syndrome or a predominant cardiovascular symptomatology characterized by endothelial dysfunction-microangiopathy, myocarditis, thrombosis and myocardial infarction (Figure 3).
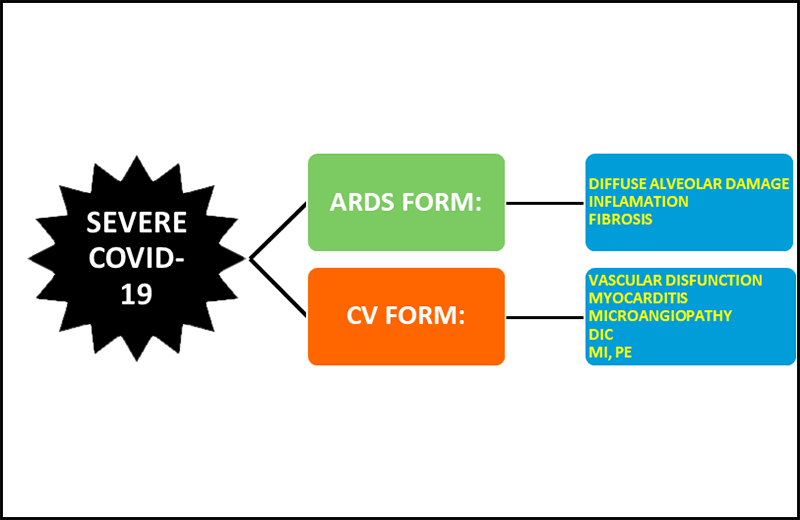
Figure 3
Figure 3
Severe Covid-19. This disease presents as an ARDS or a cardiovascular predominant form.
Therefore, the therapeutic protocol should be individualized based on individual's risk for developing either a purely respiratory form of Covid-19 or the ACE2 dependent cardiovascular Covid-19.
Future research focusing on finding an effective therapy to tackle SARS-CoV2 should concentrate in verifying the most promising experimental studies in animal model and if possible, in Covid-19. Based on the current evidence, most likely a curative solution could be revealed by studies evaluating the SARS-CoV2-ACE2 interaction, morphology of the S-protein and its activators as well as by research investigating the immunological response to SARS-CoV2 including the anti-viral role of plasmacytoid dendritic cells and anti-inflammatory reprogramming of macrophages (Table 1).
Table 1
| RESEARCH FOCUS | NOTES | REFERENCES | ||
| SARS-CoV2-S protein RBD structure | The compact structure of S protein RBD allows an efficient attachment of SARS-CoV2 to ACE2. More experimental and clinical studies should be conducted in Covid-19 patients. | Shang et al (2020) | ||
| SARS-CoV2-S protein furin-like cleavage | The furin-like cleavage acts as a switch-on mechanism for SARS-CoV2 enhancing its intrusion into cells. More experimental and clinical studies should be conducted in Covid-19 patients. | Coutard et al (2020) | ||
| TMPRSS2 | This transmembrane protein serine 2 is used for priming of SARS-CoV2 S protein. Camostate mesylate which blocks TMPRSS2 is already in use in pancreatitis and drug induced injury of lung. | Hoffmann et al (2020) | ||
| SARS-CoV2-D614G mutation | SARS-CoV2 bids with increased efficiency to cells displaying ACE2 if the D614G mutation is present. To date this have not been investigated in animal models or Covid-19 patients. | Zhang et al (2020) | ||
| IL1, IL6, TNF, IRF3, IRF7, AP-1, ATF-2/jun, Nf-kB, TLr-3, TLR-7, CXCL10, nsp1, nsp3, nsp7, nsp15, ORF3b, ORF3b, ORF9b | These are potential modulators of anti SARS-CoV2 response. Their action should be assessed in more experimental and clinical studies conducted in Covid-19 patients. | Dosch et al (2009), Felsenstein et al (2020), Russell et al (2020), DeDiego et al (2014), de Wit al (2016), Züst et al 2007), Frieman et al (2009) | ||
| Ace2-Ang1-7-Mas interaction | The action of ACE2-Ang1-7 on Mas receptors located on macrophages induces an anti-inflammatory reprogramming. This finding should be verified in animal models of SARS-CoV2 infection and Covid-19 patients. | de Carvalho Santuchi et al (2019) | ||
| SARS-CoV-papain-like protease | This factor inhibits IRF 3 phosphorylation decreasing endogenous interferon. This finding should be verified in animal models of SARS-CoV2 infection and Covid-19 patients. | Matthews et al (2014) | ||
| Plasmacytoid dendritic cells are capable to manufacture interferon during SARS-CoV infection | This finding should be verified in animal models of SARS-CoV2 infection and Covid-19 patients | Cervantes-Barragan et al (2006), Scheuplein et al (2015) | ||
| SARS-CoV delay in interferon production | This finding should be verified in animal models of SARS-CoV2 infection and Covid-19 patients | Kindler and Thiel (2016) | ||
Acknowledgements
Competing interests
The authors declare no conflict of interest.
Original submitted files for images
Below are the links to the authors’ original submitted files for images.
Original image file for Figure 1
{kind=link}
Original image file for Figure 2
{kind=link}
Original image file for Figure 3
{kind=link}
References
- Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007 Oct;20(4):660-94. doi:10.1128/CMR.00023-07
- Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004 Jun;203(2):631-7. doi:10.1002/path.1570
- Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev. 2008 Sep;72(3):457-70. doi:10.1128/MMBR.00004-08
- Lessons from SARS: a new potential therapy for acute respiratory distress syndrome (ARDS) with angiotensin converting enzyme 2 (ACE2). Masui. 2008 Mar;57(3):302-10.
- Angiotensin-converting enzyme-2: a molecular and cellular perspective. Cell Mol Life Sci. 2004 Nov;61(21):2704-13. doi:10.1007/s00018-004-4240-7
- Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. 2020;383(2):120-128. doi:10.1056/NEJMoa2015432
- Angiotensin II signal transduction in vascular smooth muscle: pathways activated by specific tyrosine kinases. J Am Soc Nephrol. 1999 Jan;10 Suppl 11:62-8. doi:10.1161/01.res.80.5.607
- Role of ACE inhibition or AT1 blockade in the remodeling following myocardial infarction. Basic Res Cardiol. 1998;93 Suppl 2:92-100. doi:10.1007/s003950050228
- Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005 Feb;26(4):369-75. doi:10.1093/eurheartj/ehi114
- Evolution of Angiotensin Peptides and Peptidomimetics as Angiotensin II Receptor Type 2 (AT2) Receptor Agonists. Biomolecules. 2020.10(4) doi:10.3390/biom10040649
- Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007 Jan;292(1):82-97. doi:10.1152/ajpcell.00287.2006
- Transactivation of the insulin-like growth factor-I receptor by angiotensin II mediates downstream signaling from the angiotensin II type 1 receptor to phosphatidylinositol 3-kinase. Endocrinology. 2004 Jun;145(6):2978-87. doi:10.1210/en.2004-0029
- Angiotensin II regulates vascular and endothelial dysfunction: recent topics of Angiotensin II type-1 receptor signaling in the vasculature. Curr Vasc Pharmacol. 2006 Jan;4(1):67-78. doi:10.2174/157016106775203126
- Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal. 2013;19(10):1110-20. doi:10.1089/ars.2012.4641
- Angiotensin II limits NO production by upregulating arginase through a p38 MAPK-ATF-2 pathway. Eur J Pharmacol. 2015;746:106-14. doi:10.1016/j.ejphar.2014.10.042
- ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors. Can J Physiol Pharmacol. 2002 Apr;80(4):346-53. doi:10.1139/y02-021
- A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238-43. doi:10.1074/jbc.M002615200
- Membrane-associated zinc peptidase families: comparing ACE and ACE2. Biochim Biophys Acta. 2005;1751(1):2-8. doi:10.1016/j.bbapap.2004.10.010
- Angiotensin-converting enzyme 2: a functional receptor for SARS coronavirus. Cell Mol Life Sci. 2004 Nov;61(21):2738-43. doi:10.1007/s00018-004-4242-5
- Angiotensin-converting enzyme-2: a molecular and cellular perspective. Cell Mol Life Sci. 2004 Nov;61(21):2704-13. doi:10.1007/s00018-004-4240-7
- The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020 Jun;76:14-20. doi:10.1016/j.ejim.2020.04.037
- A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87(5):1-9. doi:10.1161/01.res.87.5.e1
- Angiotensin-(1-7) formation in the intact human heart: in vivo dependence on angiotensin II as substrate. Circulation. 2003;108(14):1679-81. doi:10.1161/01.CIR.0000094733.61689.D4
- Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277(17):14838-43. doi:10.1074/jbc.M200581200
- MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am J Physiol Cell Physiol. 2008 Nov;295(5):1169-74. doi:10.1152/ajpcell.00145.2008
- The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol Rev. 2018;98(1):505-553. doi:10.1152/physrev.00023.2016
- Aldosterone, but not angiotensin II, reduces angiotensin converting enzyme 2 gene expression levels in cultured neonatal rat cardiomyocytes. Circ J. 2008 Aug;72(8):1346-50. doi:10.1253/circj.72.1346
- Activation of the MAS receptor by angiotensin-(1-7) in the renin-angiotensin system mediates mesenteric vasodilatation in cirrhosis. Gastroenterology. 2013 Oct;145(4):874-884. doi:10.1053/j.gastro.2013.06.036
- Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013.216(2) doi:10.1530/JOE-12-0341
- Counter-regulatory effects played by the ACE - Ang II - AT1 and ACE2 - Ang-(1-7) - Mas axes on the reactive oxygen species-mediated control of vascular function: perspectives to pharmacological approaches in controlling vascular complications. Vasa. 2014 Nov;43(6):404-14. doi:10.1024/0301-1526/a000387
- Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100(14):8258-63. doi:10.1073/pnas.1432869100
- The Vasoactive Mas Receptor in Essential Hypertension. J Clin Med. 2020.9(1) doi:10.3390/jcm9010267
- Activation of the ACE2/Ang-(1-7)/Mas pathway reduces oxygen-glucose deprivation-induced tissue swelling, ROS production, and cell death in mouse brain with angiotensin II overproduction. Neuroscience. 2014;273:39-51. doi:10.1016/j.neuroscience.2014.04.060
- Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450-4. doi:10.1038/nature02145
- Immunohistochemical, in situ hybridization, and ultrastructural localization of SARS-associated coronavirus in lung of a fatal case of severe acute respiratory syndrome in Taiwan. Hum Pathol. 2005 Mar;36(3):303-9. doi:10.1016/j.humpath.2004.11.006
- SARS-CoV replication and pathogenesis in an in vitro model of the human conducting airway epithelium. Virus Res. 2008 Apr;133(1):33-44. doi:10.1016/j.virusres.2007.03.013
- Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol. 2010 Jan;84(2):1198-205. doi:10.1128/JVI.01248-09
- Origin, Potential Therapeutic Targets and Treatment for Coronavirus Disease (COVID-19). Pathogens. 2020.9(4) doi:10.3390/pathogens9040307
- Renin-angiotensin system at the heart of COVID-19 pandemic. Biochimie. 2020 Jul;174:30-33. doi:10.1016/j.biochi.2020.04.008
- COVID-19 diagnosis and management: a comprehensive review. J Intern Med. 2020 Aug;288(2):192-206. doi:10.1111/joim.13091
- COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res. 2020. doi:10.1093/cvr/cvaa106
- Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020 May;581(7807):221-224. doi:10.1038/s41586-020-2179-y
- The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. 2020 Apr.176 doi:10.1016/j.antiviral.2020.104742
- SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271-280. doi:10.1016/j.cell.2020.02.052
- The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv. June 2020.:148726-. doi:10.1101/2020.06.12.148726
- Smoking Upregulates Angiotensin-Converting Enzyme-2 Receptor: A Potential Adhesion Site for Novel Coronavirus SARS-CoV-2 (Covid-19). J Clin Med. 2020.9(3) doi:10.3390/jcm9030841
- Association of respiratory allergy, asthma, and expression of the SARS-CoV-2 receptor ACE2. J Allergy Clin Immunol. 2020 Jul;146(1):203-206. doi:10.1016/j.jaci.2020.04.009
- Covid-19, the kidney and hypertension.. Harefuah. 2020 Apr;159(4):231-234.
- Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res. 2020 Jul;43(7):648-654. doi:10.1038/s41440-020-0455-8
- Risks of ACE Inhibitor and ARB Usage in COVID-19: Evaluating the Evidence. Clin Pharmacol Ther. 2020 Aug;108(2):236-241. doi:10.1002/cpt.1863
- [ACE-inhibitors, angiotensin receptor blockers and severe acute respiratory syndrome caused by coronavirus]. G Ital Cardiol (Rome). 2020 May;21(5):321-327. doi:10.1714/3343.33127
- Substituting Angiotensin-(1-7) to Prevent Lung Damage in SARS-CoV-2 Infection?. Circulation. 2020;141(21):1665-1666. doi:10.1161/CIRCULATIONAHA.120.047297
- Angiotensin-(1-7) and Alamandine Promote Anti-inflammatory Response in Macrophages In Vitro and In Vivo. Mediators Inflamm. 2019.2019 doi:10.1155/2019/2401081
- SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009 Jun;142(1-2):1-2. doi:10.1016/j.virusres.2009.01.005
- COVID-19: Immunology and treatment options. Clin Immunol. 2020 Jun.215 doi:10.1016/j.clim.2020.108448
- Associations between immune-suppressive and stimulating drugs and novel COVID-19-a systematic review of current evidence. Ecancermedicalscience. 2020.14 doi:10.3332/ecancer.2020.1022
- Inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014;88(2):913-924. doi:10.1128/JVI.02576-13
- SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016 Aug;14(8):523-34. doi:10.1038/nrmicro.2016.81
- SARS-CoV and IFN: Too Little, Too Late. Cell Host Microbe. 2016;19(2):139-41. doi:10.1016/j.chom.2016.01.012
- The SARS coronavirus papain like protease can inhibit IRF3 at a post activation step that requires deubiquitination activity. Virol J. 2014.11 doi:10.1186/s12985-014-0209-9
- Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 2007.3(8) doi:10.1371/journal.ppat.0030109
- Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83(13):6689-6705. doi:10.1128/JVI.02220-08
- Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109(3):1131-7. doi:10.1182/blood-2006-05-023770
- High secretion of interferons by human plasmacytoid dendritic cells upon recognition of Middle East respiratory syndrome coronavirus. J Virol. 2015 Apr;89(7):3859-69. doi:10.1128/JVI.03607-14
- Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020. doi:10.1111/his.14134
- Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta. 2020 Aug;507:167-173. doi:10.1016/j.cca.2020.04.027
- Raised plasma renin and prorenin in rheumatoid vasculitis. Ann Rheum Dis. 1990 Jul;49(7):517-20. doi:10.1136/ard.49.7.517
- The renin-angiotensin system as a primary cause of polyarteritis nodosa in rats. J Cell Mol Med. 2010 Jun;14(6A):1318-27. doi:10.1111/j.1582-4934.2009.00778.x
- Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke. 2012 Dec;43(12):3358-63. doi:10.1161/STROKEAHA.112.667063
- Early life stress sensitizes rats to angiotensin II-induced hypertension and vascular inflammation in adult life. Hypertension. 2010 Feb;55(2):494-9. doi:10.1161/HYPERTENSIONAHA.109.145391
- SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009 Jul;39(7):618-25. doi:10.1111/j.1365-2362.2009.02153.x
- Cell type-specific expression of the putative SARS-CoV-2 receptor ACE2 in human hearts. Eur Heart J. 2020;41(19):1804-1806. doi:10.1093/eurheartj/ehaa311
- Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm. 2020. doi:10.1016/j.hrthm.2020.05.001
- Fatal Eosinophilic Myocarditis in a Healthy 17-Year-Old Male with Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2c). Fetal and pediatric pathology. 2020;39(3):263-268. doi:10.1080/15513815.2020.1761491
- Acute myocarditis presenting as a reverse Tako-Tsubo syndrome in a patient with SARS-CoV-2 respiratory infection. Eur Heart J. 2020;41(19):1861-1862. doi:10.1093/eurheartj/ehaa286
- Myocarditis in a patient with COVID-19: a cause of raised troponin and ECG changes. Lancet. 2020.395(10235) doi:10.1016/S0140-6736(20)30912-0
- SARS-CoV-2 receptor ACE2 expression in the human heart: cause of a post-pandemic wave of heart failure?. Eur Heart J. 2020;41(19):1807-1809. doi:10.1093/eurheartj/ehaa410
- COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033-2040. doi:10.1182/blood.2020006000
- COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020;75(23):2950-2973. doi:10.1016/j.jacc.2020.04.031
- Intra-arterial thrombosis associated with COVID-19. Journal of vascular surgery. May 2020;S0741-5214(20):31235-0. doi:10.1016/j.jvs.2020.05.019
- Prothrombotic phenotype in COVID-19 severe patients. Intensive Care Med. 2020;46:1502-1503. doi:10.1007/s00134-020-06082-7
- First case of Covid-19 presented with cerebral venous thrombosis: A rare and dreaded case. Rev Neurol (Paris). 2020 Jun;176(6):521-523. doi:10.1016/j.neurol.2020.04.013
- Concomitant acute aortic thrombosis and pulmonary embolism complicating COVID-19 pneumonia. Diagn Interv Imaging. 2020 May;101(5):321-322. doi:10.1016/j.diii.2020.04.003
- Analysis of complement deposition and viral RNA in placentas of COVID-19 patients. Ann Diagn Pathol. 2020.46 doi:10.1016/j.anndiagpath.2020.151530
- Elevated Troponin in Patients With Coronavirus Disease 2019: Possible Mechanisms. J. Card. Fail. 2020;26(6):30357-2. doi:10.1016/j.cardfail.2020.04.009
- Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: implications for future therapeutic directions. Clin Sci (Lond). 2012 Dec;123(11):649-58. doi:10.1042/CS20120162
- Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: potential implications for inflammation and plaque instability. Circulation. 2000;101(12):1372-8. doi:10.1161/01.cir.101.12.1372
- A novel role for calpain in the endothelial dysfunction induced by activation of angiotensin II type 1 receptor signaling. Circ Res. 2011;108(9):1102-11. doi:10.1161/CIRCRESAHA.110.229393
- Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front Physiol. 2014.5 doi:10.3389/fphys.2014.00396
- Angiotensin II and inflammation: the effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J Hum Hypertens. 2007 Jan;21(1):20-7. doi:10.1038/sj.jhh.1002101

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.