The long and winding road: detecting and quantifying Notch activation in endothelial cells
Vascular Cell. 2021;
Received: 11 March 2021 | Accepted: 10 September 2021 | Published: 20 December 2021
Vascular Cell ISSN: 2045-824X
Abstract
The family of Notch proteins plays a key role in cell fate determination. Additionally, Notch proteins regulate critical functions of the endothelium, as well as other recruited supporting cells, in concert with other pathways. Despite significant advances in the field and extensive studies focused on elucidating this pathway, many questions remain regarding Notch activation and its upstream/downstream effects, with vascular biology constituting one area of particular interest. Here, we provide a brief description of the components and functions of the Notch pathway in vasculature, followed by a detailed compilation of recommended methods of evaluation in vitro and in vivo. We provide a rationale for key elements when choosing different approaches and controls, strengths and limitations, and essential considerations when providing a meaningful interpretation of results. Our aim is to describe a careful approach to assessing Notch function in endothelial cells, based on underlying principles, with the overall goal of obtaining physiologically relevant information that will enhance our understanding of this pathway and its role in vascular biology.
Keywords
Notch retina endothelial cell gamma secretase inhibitors human umbilical vein endothelial cells HUVECIntroduction
The Notch pathway plays a number of essential roles within the field of vascular biology: it is critical for arterial differentiation, maturation, and quiescence, as well as cardiovascular function, among other processes. Notch interacts with a variety of other signaling pathways, feedback loops, and local transcription factors (for a more in-depth review of this topic, see [1]). As such, defects in the pathway are implicated in multiple pathological conditions. These include, but are not limited to, tumor angiogenesis and diseases we will describe in more detail, such as Alagille syndrome. The potential for cell type-specific downstream targets, then, can complicate the study of Notch activation within a particular system, organ, or pathology. Intriguingly, Notch activation in the form of constitutively-activated receptors often yields similar phenotypes to Notch knockouts, suggesting that dosage and cell context are critical. Adding to this complexity, much of our understanding of Notch has been derived from inhibition studies, namely Notch decoys, inhibitors, silencing techniques, and knockout models. Understanding the effects of physiological levels of Notch activation, therefore, has been elusive.
The aim of this review is to collate and summarize methods to effectively detect and quantify Notch activation. We begin with a brief description of the components of the Notch pathway before moving on to a compilation of appropriate methods for interrogation. While our list is not exhaustive, we discuss an array of widely available molecular tools for evaluating Notch activation within endothelial cells (ECs) in vitro and in murine models. We provide some examples of the utility of these methods with specific attention regarding endothelial cells. Special attention will be paid to selecting positive and negative controls, as well as their use in the proposed experiments, and the limitations of each assay. Finally, taking these elements into account, we discuss strategies for analysis and interpretation of results.
Notch Receptors
Notch is a heterodimeric, single-pass transmembrane receptor with an extracellular binding domain (NECD) and an intracellular signaling domain (NICD). The NECD consists of 29-36 epidermal growth factor (EGF)-like repeats, a negative regulatory region (NRR) which consists of three Lin-12/Notch repeats (LNR), and a heterodimerization domain. Through glycosylation, O-linked carbohydrate modifications can be attached to the EGF-like repeats in the NECD and modify Notch activation. The NICD consists of a recombination binding protein-Jκ-associated molecule (RAM), two nuclear localization signals (NLS), ankyrin repeats, a variable transactivation domain, and a proline, glutamic acid, serine, and threonine (PEST) sequence [2].
Mammals express Notch receptors 1-4. In mice, Notch1 deletion leads to embryonic lethality due to cardiovascular defects and failure to close the neural tube [3, 4]. Although Notch4 itself is not essential during development, loss of Notch4 exacerbates the severity of Notch1 mutations, [4, 5] suggesting that Notch4 has overlapping functions with Notch1 and is also involved in angiogenic development. Mutations in the Notch1 gene have been implicated in cardiac disease and improper vascular smooth muscle cell differentiation in humans [6, 7, 8]. Notch3 expressed in mural cells recruits these to the endothelium, regulating vascular leakiness and pruning [9, 10, 11]. In humans, mutations in the Notch3 gene cause Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [9], characterized by loss of pericyte coverage of brain arteriole endothelium, leading to ischemic strokes. Other disorders associated with Notch mutations include Adams-Oliver, Alagille syndrome, spondylocostal dysostosis, and Lehman syndrome (reviewed in [12]).
Notch Ligands
Mammals express Notch ligands Jagged (Jag) 1-2 and Delta-like (Dll) 1 and 3-4. Structurally, Jag and Dll are transmembrane proteins composed of an N-terminal Module at the N-terminus of Notch Ligands domain, a Delta/Serrate/Lag-2 (DSL) domain, and EGF-like repeats. In comparison to Dll, Jag ligands have an additional cysteine-rich domain [13]. Both Dll and Jag ligands act by binding to the EGF-like repeats of the NECD [14, 15].
The role of Dll4 in angiogenesis and vascular biology continues to be extensively studied. Dll4 dosage is critical during developmental and pathological angiogenesis, and heterozygous Dll4 deletions in mice mirror the phenotype of Notch1 knockouts [16, 17]. Additionally, Dll4 deletion increases vascular density [18, 19, 20]. During angiogenesis, endothelial Dll4 expression determines the number of tip cells, thus regulating the number of developing sprouts and vascular density. Endothelial Jag1, on the other hand, has the opposite effect: its deletion significantly decreases the number of retinal sprouts [21]. An independent mouse model with endothelial-specific deletion of Jag1 leads to embryonic lethality in mice due to cardiovascular defects, including dilated blood vessels and hemorrhage [22]. Furthermore, the expression of vascular smooth muscle markers is severely diminished in the knockout mice. However, no difference in apoptosis or proliferation is seen, suggesting that endothelial Jag1 is required for recruitment or differentiation of smooth muscle cells [22]. Data from both mouse models and clinical studies suggests that Jag1 defines arterial development and differentiation through vascular smooth muscle cell recruitment to ECs [21, 22, 23, 24, 25, 26, 27]. In humans, Jag1 deletions or mutations lead to Alagille syndrome, characterized by intrahepatic cholestasis and abnormalities of the heart, eye, and vertebrae [23, 24].
While deletion and knockout studies of both ligands and receptors have long been used to reveal the role of Notch in embryonic development, mouse models with conditional or constitutive activation of Notch receptors under specific promoters continue to emerge (reviewed here [28]).The study of these mouse models reveals that both Notch knockout and constitutive activation can lead to comparable, if not identical, vascular phenotypes, as in the case of early embryonic angiogenesis. This suggests that Notch activation is carefully modulated, and slight changes in activation can have profound effects on downstream targets and phenotypes.
The Notch pathway
The Notch pathway is critical during multiple stages of embryonic development, including neural tube closure and somite formation. Given Notch’s complex interactions with other local signaling pathways, it has been implicated in multiple pathologies, including vascular abnormalities. Downregulation of Notch signaling can result in cardiac defects, such as cardiac valve calcification and Alagille syndrome [23, 24, 29]. Finally, activating mutations of Notch have been linked to tumor formation, such as T-lymphocyte leukemia, breast cancer [30], and infantile hemangiomas [31, 32, 33].
Figure 1 illustrates the Notch pathway from the perspective of the receptor-expressing cell. During post-translational transport, the Notch receptor undergoes oxygen-linked glycosylation and fucosylation in the endoplasmic reticulum (Fig. 1A, step 1). Then, the Notch precursor is cleaved by a furin-like convertase within the trans-golgi network at site S1 to produce two subunits that associate to form the heterodimeric receptor [34, 35]. The receptor is then localized to the plasma membrane (Fig. 1A, step 2).
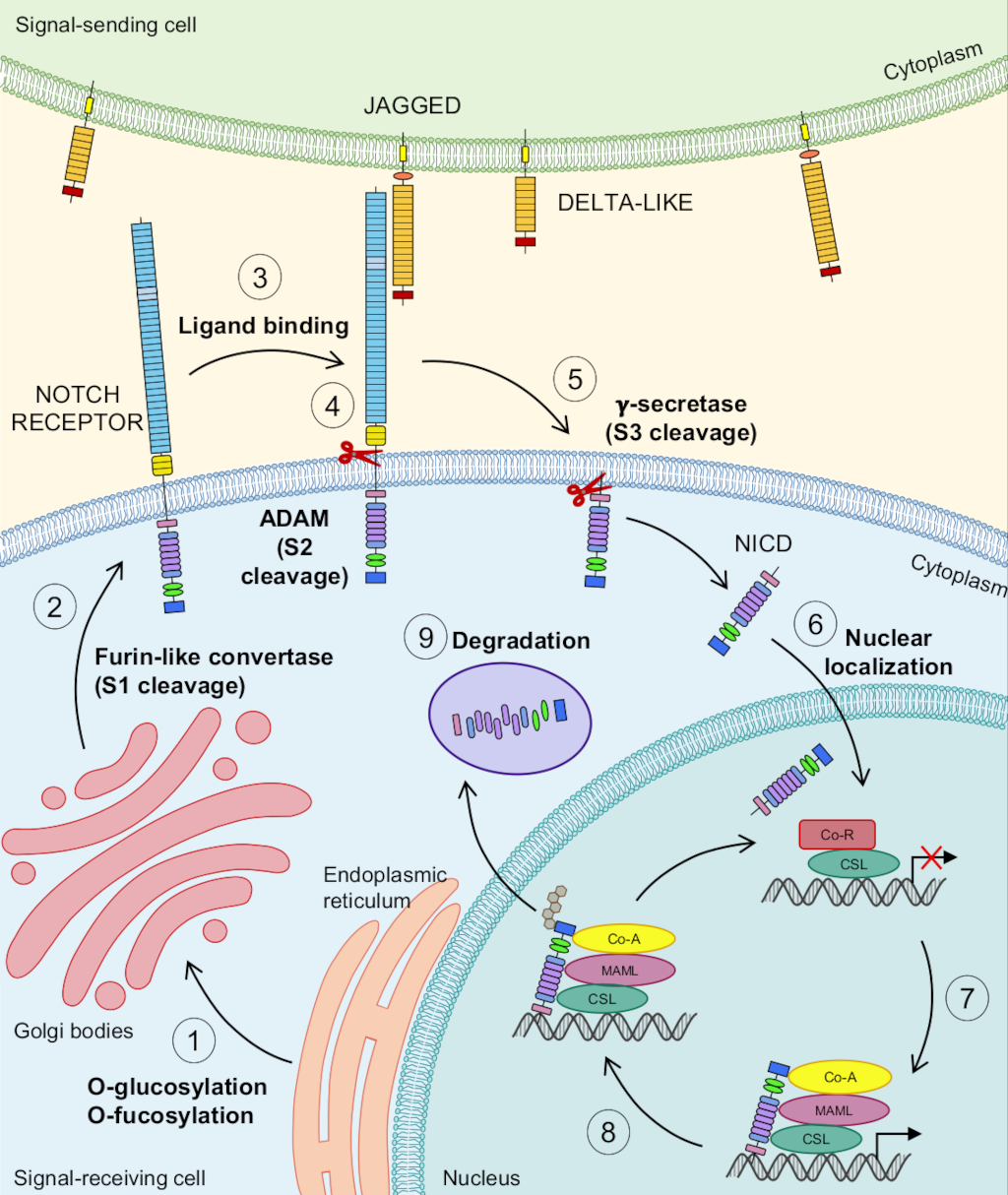
Figure 1: The Notch pathway.
Figure 1: The Notch pathway.
1) Following translation, the Notch receptor undergoes oxygen-linked glycosylation and fucosylation in the endoplasmic reticulum. 2) Notch is then transported to the Golgi bodies for further modification. Within the Golgi bodies, a furin-like convertase cleaves the Notch receptor at S1 before it is transported to the plasma membrane. 3) Two ligand classes can bind to and activate Notch: Jagged and Delta-like. 4) Upon ligand binding, Notch is cleaved by a protease in the A Disintegrin and Metalloprotease domain-containing protein (ADAM) family at S2, either ADAM10 and/or ADAM17. 5) The protein complex γ-secretase performs a subsequent cleavage at S3 (valine 1744 in both human and murine Notch1), releasing the intracellular domain of the Notch receptor (NICD) into the cytoplasm. 6) The NICD translocates to the nucleus. 7) In the nucleus, the NICD binds to the DNA-binding transcription factor CBF1, suppressor of hairless, Lag-1 (CSL). Upon binding, the NICD will displace co-repressors and recruit transcriptional co-activators, such as Mastermind/Lag-3 (MAML). This process allows for the regulation of Notch downstream target genes. 8) The NICD is targeted for ubiquitination by the E3 ubiquitin ligase Suppressor/Enhancer of Lin-12 (SEL-10), thus targeting it for degradation. 9) The NICD is degraded by the proteasome.
Binding of Delta or Jagged ligands from adjacent signal-sending cells releases auto-inhibition and exposes the Notch receptor’s S2 cleavage site (Fig. 1A, step 3). Studies have shown that ligands need to be endocytosed via the Epsin pathway and ubiquitinated by the signal-sending cell in order for activation to take place [36, 37]. Evidence further suggests that this endocytosis applies the pulling force necessary for Notch activation to occur ([38] and reviewed in [39]). Ligands expressed in the same cell as the receptor can bind to receptors (in cis), however, inhibition results from this interaction (reviewed in [40]). Recent evidence has further elucidated that cis-interactions are initially activating but ultimately become inhibiting as ligand levels increase [41].
S2 cleavage is carried out by members of A Disintegrin And Metalloproteases (ADAM), a family of transmembrane enzymes with multiple known targets (Fig. 1A, step 4) [42]. Studies suggest that ADAM10 is the main protease regulating Notch1 S2 cleavage, but there is residual activity by ADAM17/TACE [43, 44, 45, 46, 47, 48, 49]. Additional research has suggested that ADAM10 is involved in ligand-dependent activation of the Notch pathway whereas ADAM17/TACE activation is ligand-independent and insufficient to rescue Notch activation in the absence of ADAM10 [50].
S2 cleavage is immediately followed by a third cleavage at S3 (valine 1744 in both human and murine Notch1, Fig. 1A, step 5) mediated by presenilin, a transmembrane protease in the γ-secretase protein complex [51, 52, 53]. This S3 cleavage releases the NICD from the membrane, allowing it to translocate into the nucleus due to its two NLS domains (Fig. 1A, step 6). Once in the nucleus, the NICD associates with the DNA-binding transcription factor CBF1, suppressor of hairless, Lag-1 (CSL) and RAM complex [54]. This complex then displaces co-repressors and recruits co-activators, such as Mastermind/Lag-3 (MAML) [55], leading to downstream gene transcription and corresponding biological effects. Activation or repression of target genes by Notch is cell-context dependent (reviewed in [1]), and certain Notch targets can function as repressors themselves, amplifying the downstream effects [39, 56, 57, 58].
Following ligand binding, the NECD is endocytosed by the ligand-expressing cell [59]. Within the receptor-expressing cell, the NICD is later ubiquitinated by E3 ligase Suppressor/Enhancer of Lin-12 (SEL-10) [60] and degraded by proteasomes (Fig. 1A, steps 8 and 9).
NOTCH ACTIVATION IN VITRO
Next, we will discuss the advantages and limitations of various assays and methods available to detect and quantify Notch activation in vitro, beginning with constructs and compounds that can serve as positive controls or inhibitors.
Use of Ligands
Although secreted ligands have been shown to activate Notch in C. elegans [61], Klose et al. provide evidence that soluble ligands (not fixed to a matrix) inhibit Notch during angiogenesis, both in vivo and in vitro [62]. Varnum-Finney et al. demonstrated that ligands can activate Notch if they are fixed to plastic, even if they are not able to apply a pulling force on the receptor, while confirming that these same ligands inhibit Notch activation if they are in suspension [63]. Therefore, pre-fixing ligands to a plate, before plating cells, can provide a useful experimental approach for inducing Notch activation in vitro. It should be stressed, however, that soluble ligands should not be used to activate Notch in vitro, as this will result in inhibition. Figure 2 illustrates how to use coated ligands to activate Notch in vitro. Lastly, overexpressing ligands by transfection can also be used for interrogating Notch activation. Conversely, eliminating them via an approach such as siRNA can be used as a negative control.
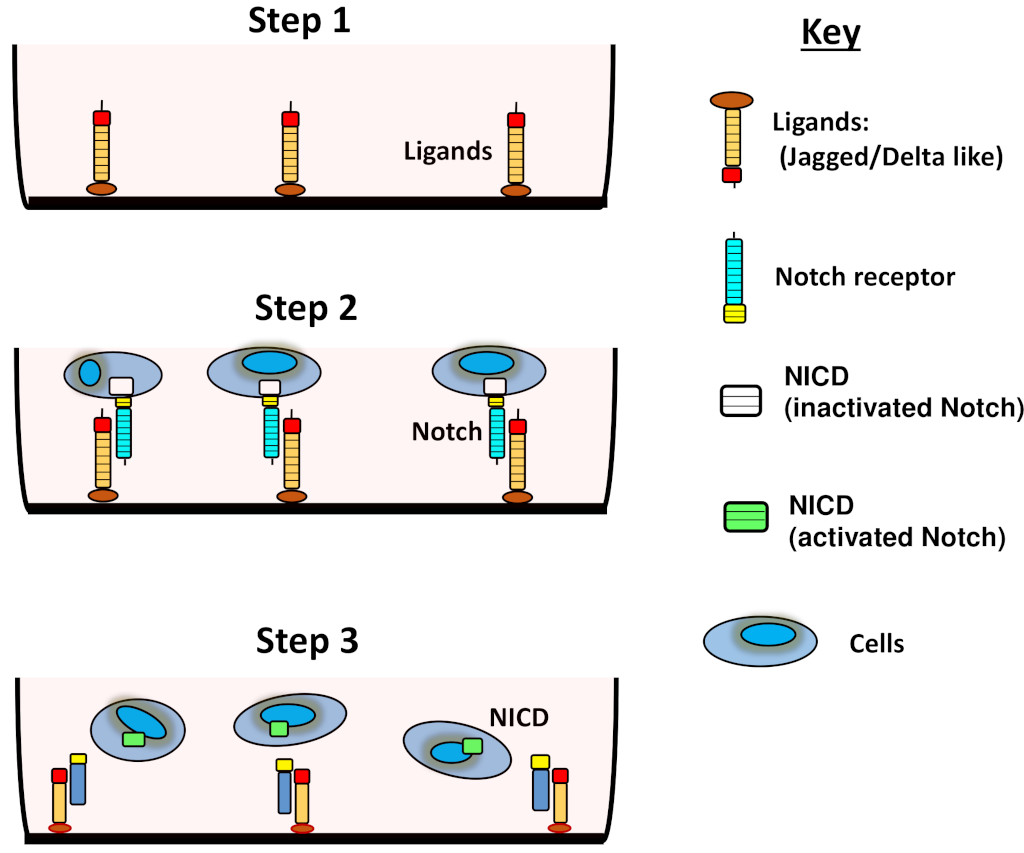
Figure 2: Activating Notch with soluble ligands.
Figure 2: Activating Notch with soluble ligands.
(1) Coating the surface of plates and fixing ligands prior to (2) seeding cells will result in (3) Notch activation (cleavage of the NICD) and its translocation to the cell nucleus. This assay can be performed with both non-adherent cells, as depicted, or adherent cells (not shown).
Positive controls: EDTA, EGTA, and plasmids.
Ethylenediaminetetraacetic acid (EDTA) and egtazic acid (EGTA) activate Notch1 by chelating the calcium ions that provide structural stability to the negative regulatory region (NRR) of the NECD [64]. The Lin-12/Notch repeats (LNR) within the NRR relies upon disulfide bonding and calcium ion coordination for structural integrity [65]. Calcium chelation by EDTA or EGTA detaches the LNR from the heterodimerization region of the NRR [64]. The LNR then adopts a more relaxed structure, thus exposing the Notch EC domain to cleavage at S2 [66]. In summary, EDTA and EGTA destabilize Notch, leading to its cleavage and activation. EDTA activation will be inhibited by γ-secretase, as demonstrated in Fig. 3. EDTA has been used as a Notch activator in multiple cell lines [46, 50, 64, 67, 68], and can be utilized as a positive control when studying Notch activation in ECs. It is important to bear in mind, however, that this activation will supersede physiological levels.
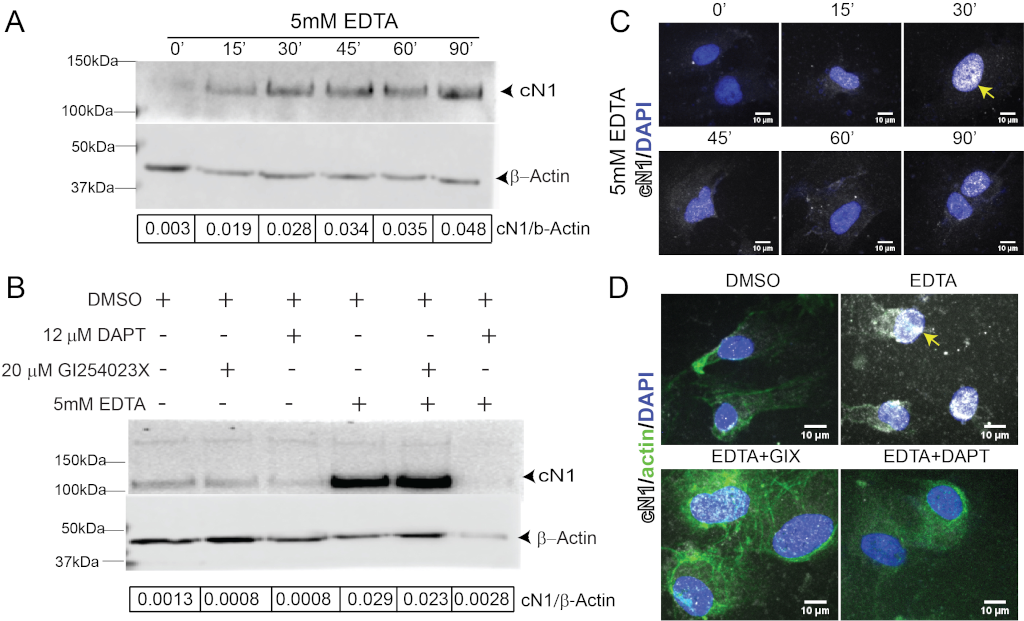
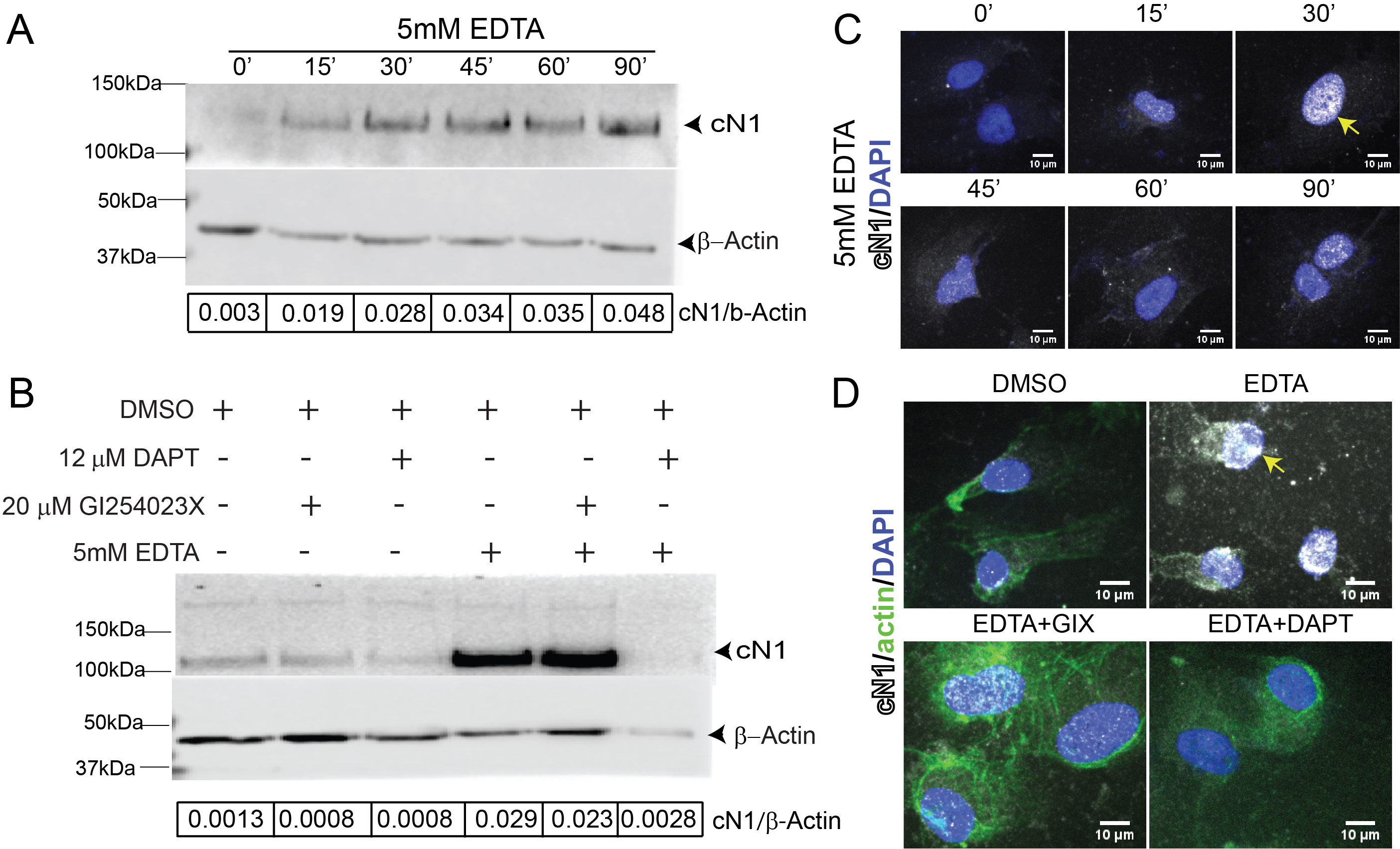
Figure 3: EDTA is a positive control for Notch activation in HUVEC.
Figure 3: EDTA is a positive control for Notch activation in HUVEC.
A) Cleaved Notch1 (cN1, upper panel, and beta-actin, lower panel, with ratio of cN1 to Actin in the lower bar) increased as early as 15 minutes after EDTA stimulation (lane 2) compared to untreated controls (lane 1) and is sustained for at least 90 minutes (lane 6). B) The ADAM10 inhibitor GIX reduces baseline cN1 compared to dimethyl sulfoxide (DMSO) controls (lane 2 vs lane 1); the γ-secretase inhibitor DAPT equally reduces cN1 (lane 3 vs lane 1). GIX does not prevent cN1 activation by EDTA (lane 4 vs 5), while DAPT completely abrogates Notch1 cleavage after EDTA stimulation (lane 6 vs 4). C) Immunocytochemistry shows cN1 (white) nuclear localization increases after EDTA stimulation, peaking at 30 minutes (yellow arrow) before a second increase at 90 minutes (bottom right panel). D) Immunocytochemistry reveals that 30 minutes after EDTA stimulation, cN1 localizes to the nucleus (yellow arrow). This is abrogated by DAPT (bottom right panel, loss of white staining), but not by GIX (bottom left panel, residual white staining). EDTA rearranges β-actin filaments (upper right, green), altering HUVEC morphology.
Additionally, EDTA can also cause cells to detach from plate surfaces. Detachment can affect assays, such as SDS-PAGE and quantitative real-time PCR (qPCR), because it may reduce the total amount of protein or RNA collected. EDTA can also lead to the rearrangement of F-actin filaments. As a result, HUVECs may lose their characteristic cobblestone appearance following EDTA stimulation, thereby altering imaging. Such morphological changes may persist for a few hours [49].
Alternatively, multiple plasmids exist for constitutively activated forms of the Notch receptors, such as N1ICD and Int3/Notch4 [51, 69]. These can be transfected into ECs in order to interrogate their effects. Cell behavior, however, may be dramatically altered in some cases; for example, N1ICD arrests endothelial cell proliferation [70], rendering this method challenging when propagating cells or performing certain assays. Many of these plasmids are commercially available but may have differing structures; for example, some preserve the S3 site while others do not. Multiple post-translational modifications such as phosphorylation, acetylation, methylation and ubiquitination, affect the transcriptional activity of Notch. Thus, constructs lacking the S3 site may also lack these post-translational modifications which could result in altered downstream target activation compared to endogenous Notch.
ADAM, γ-secretase and Decoy inhibitors.
Ludwig et al. reported the IC50 values of a number of viable metalloprotease inhibitors for ADAM10 and ADAM17/TACE [71]. Among these, our group uses G1254023X (GIX). Both ADAM10 and ADAM17/TACE can be inhibited by GIX, but at certain concentrations GIX can be used as a selective inhibitor for ADAM10 in vitro due to its more than 100-fold higher affinity for ADAM10 than for ADAM17 [71].
There are many selective γ-secretase inhibitors (GSIs) that can be used both in vitro and in vivo, such as Compound E and Dibenzazepine (DBZ), N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) [72], with DAPT being the most widely used GSI in laboratories [73]. Empirically, we have found that both GIX and DAPT require overnight (8-16 hour) incubations prior to testing activation to effectively inhibit Notch in vitro. Other mechanisms to inhibit Notch are Notch Decoys [74], which prevent activation by competing for ligand specific binding sites. Another mechanism to inhibit Notch activation are neutralizing antibodies [20, 75]. All of these can be used in vitro and in vivo.
Antibodies for detecting Notch activation in vitro.
Careful consideration should be taken when choosing antibodies to quantify Notch activation. A cleaved Notch1 (cN1) antibody (Cell Signaling Technology, Danvers, MA, USA) that only detects the residue exposed after γ-secretase cleavage at S3, Val1744 in humans and mice, and runs at 110 kDa in SDS-PAGE is an excellent way to quantify activated Notch1, when normalized to a housekeeping control such as β-actin. Alternatively, a commercially available antibody that recognizes activated Notch1 amino acids 1755-1767 can be utilized (Abcam). This peptide sequence is only exposed after γ-secretase cleavage, making this antibody another viable way to detect Notch activation. An antibody recognizing other residues in the NICD will be able to detect three products: full length Notch (over 290 kDa, glycosylated and difficult to transfer), Notch cleaved at site S2 but not at S3 (120 kDa), and active Notch cleaved at S3 (110 kDa). Since the fragment produced after S2 cleavage differs only by about 10 kDa from the fragment produced after S3 cleavage, these latter two products are difficult to resolve with electrophoresis, making comparison between levels of cN1 and these other forms difficult. Thus, an antibody directed at Val1744, or a peptide sequence exposed only after S3 cleavage, is a more efficient and reliable way to quantify Notch activation.
Immunoblotting.
Notch1 activation can be quantified by estimating the amount of active Notch1 relative to a housekeeping protein. This is best performed using an antibody that recognizes cN1, as discussed above, and is particularly useful for identifying an appropriate time at which the Notch1 is cleaved at the S3 site and released. We found whole cell lysates of HUVECs incubated with 5 mM EDTA for 5 minutes, followed by EC media for one-hour, demonstrated strong Notch1 activation beginning at 15 minutes and persisting until 90 minutes after stimulation (Fig. 3A, lane 1 vs lanes 2-6). DAPT abrogated Notch1 activation in HUVECs 30 minutes after EDTA stimulation (Fig. 3B, lanes 4 vs 6), indicating that inhibition of Notch signaling can be detected via immunoblotting. GIX, however, failed to do so (Fig. 3B lanes 4 vs 5), confirming that other metalloprotease(s) can process Notch activation in vitro, and suggesting that GIX alone is insufficient for Notch inhibition. Thus, quantifying anti-cN1 normalized to β-actin after EDTA stimulation can be used to set up immunoblotting conditions and identify peak incubation time within the desired system.
Immunocytochemistry.
When performed appropriately, immunocytochemistry (ICC) can visualize the cleaved NICD within the nucleus or cytoplasm, which may be advantageous for analysis. Visualizing at high magnification (40x or more) using confocal microscopy aids in correct colocalization to the cell nucleus. The nuclear signal can also be quantified, by software such as ImageJ, using nuclear-colocalization tools with dyes such as 4′,6-diamidino-2-phenylindole (DAPI). A detailed section on ImageJ analysis and sources is included below in the in vivo section. When performed in concert with immunoblotting, ICC can help more clearly identify the relative quantity of NICD and its localization. This may be relevant, for example, if there is cN1 present within the cell but not localized to the nucleus; without such localization, cN1 will not be transcriptionally active.
Via ICC, we found nuclear cN1 began increasing 15 minutes after EDTA stimulation (Fig. 3C, white pseudocolor over DAPI, blue), peaked after 30 minutes, then decreased, and had a second peak 90 minutes later. This is notable when compared against detection of cN1 in whole cell lysates by immunoblotting, which found that cN1 levels increase constantly after EDTA activation. Immunocytochemistry results suggest that 30 minutes is the peak of nuclear cN1 after EDTA stimulation in HUVECs (Fig. 3C), despite the continued presence of cN1 within the cell (Fig. 3A). Nuclear cN1 increases again at 90 minutes (Fig. 3C, bottom right panel), although not to the peak levels observed at 30 minutes. DAPT elimination of Notch1 activation is detectable via ICC (Fig. 3D, upper right vs lower right panels, white). Similar to immunoblotting, GIX reduced nuclear cN1 (Fig. 3D top right vs lower left panels, white) but did not eliminate its presence.
In summary, increased whole cell lysate cN1 levels identified by immunoblotting do not necessarily translate into changes in transcriptional activation.
Enzyme-linked immunosorbent assay (ELISA).
The anti-cleaved or activated Notch1 antibodies described above can be used to set up an in-house ELISA for quantifying cN1. Alternatively, there are commercially available kits that use this technique. When setting up ELISA in-house, the lack of a positive control can be a limitation. Moreover, identifying nuclear localization is not possible. Despite these constraints, ELISA can provide useful quantitative results. For example, Notch3 activation has been assessed, both in vitro and in vivo, using an ELISA against Notch3 extracellular domain in supernatant or circulation per the protocol detailed by Primo et al [76].
Luciferase assays.
Luciferase constructs fuse the Notch CSL promoter region (with multiple CSL binding sites), to the firefly luciferase gene. Thus, luciferase is expressed whenever Notch is activated in the transfected cell. Co-transfection with a Renilla luciferase plasmid, whose expression does not depend on Notch activation (or any other gene), normalizes transfection between different wells. Luciferase assays measure Notch activation without the usage of a cN1 antibody. As such, they are an excellent confirmatory or secondary method when used with any of the assays discussed above. In Table 1, we list some luciferase constructs that can be used to detect Notch activation in vitro.
Table 1
| Plasmid Name | Catalog number | Company | Donating Investigator |
|---|---|---|---|
| 4xCSL-luciferase | 41726 | Addgene | Kopan |
| pHes1(467 RBPj(-))-luc | 43805 | Addgene | Kageyama |
| Notch1 Pathway Reporter Kit (Human) | 79503 | BPS Bioscience | - |
| Notch1 /CSL Reporter Kit | Kit-1786 | Creative Biomart | - |
We found that luciferase expression peaks between 6 to 8 hours after 5mM EDTA stimulation in HUVECs compared to Hank’s buffered saline solution (HBSS) controls (9.4±1.3 vs 74.1±26.5 Relative Luciferase/Renilla units, p<0.0001), making these appropriate time points to detect Notch activation in ECs [49]. A 6-hour time point was reported by Tiyanont et al. 2011 using U2OS cells in a comparable luciferase assay system, suggesting this may be an appropriate timepoint across different cell lines and types [66].
Quantitative real-time PCR.
The Hairy Enhancer of Split (Hes) and Hes-related with YRPW motif (Hey) families of basic-helix-loop-helix transcription effectors and repressors are frequently used as readouts for Notch activation. Hes1 and Hey1, in particular, have been extensively reported to be Notch downstream targets [51, 77, 78, 79]. Changes in transcription levels can be measured using qPCR, as evidenced by EDTA upregulation of Hes1 (12.00-fold ± 1.30, p<0.001) and Hey1 (6.71-fold ± 0.70, p<0.001) in HUVECs stimulated and incubated for one-hour in basal media (Fig. 4A).
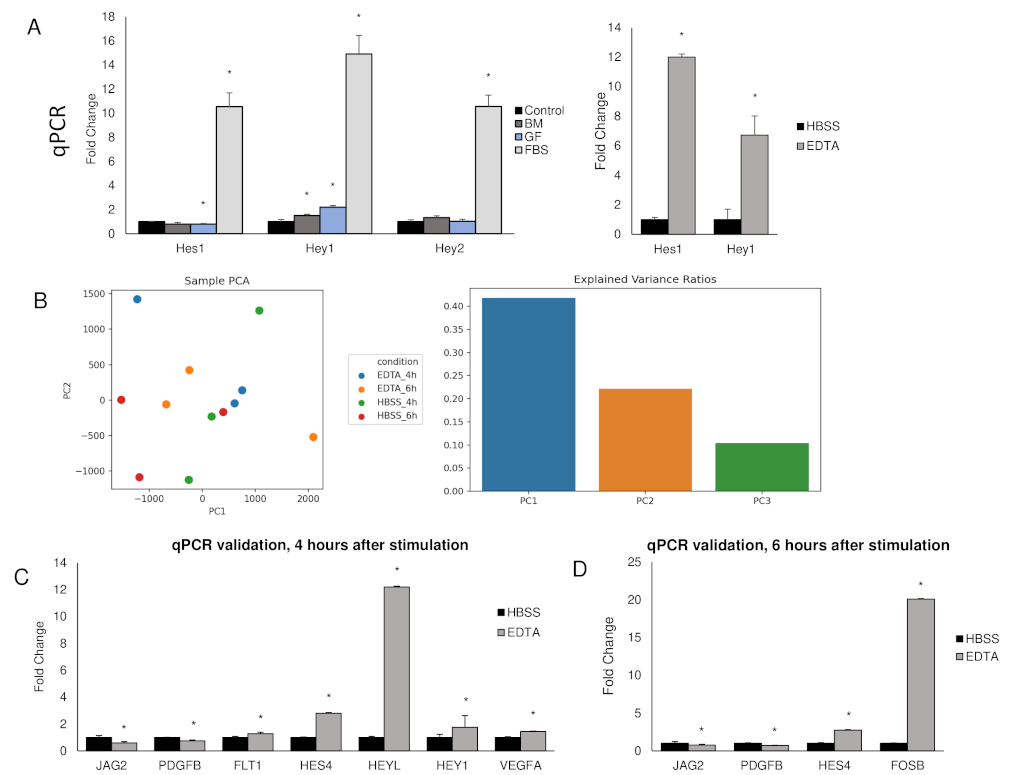
Figure 4: EDTA and fetal bovine serum (FBS) activate known Notch target genes
Figure 4: EDTA and fetal bovine serum (FBS) activate known Notch target genes
A) qPCR measuring transcription levels of canonical Notch downstream targets Hes1, Hey1, and Hey2 in HUVECs following one-hour incubation. Control samples received no media change, BM samples received basal media (no growth factors nor FBS), GF samples received basal media with growth factors, FBS samples received full media. Full media resulted in upregulation of Hes1 (10.54-fold ± 1.14), Hey1 (14.90-fold ± 1.53), and Hey2 (10.55-fold ± 0.94). Growth factors alone upregulated Hey1 (2.20-fold ± 0.14) but did not upregulate Hes1 or Hey2. Basal media alone resulted in upregulation of Hey1 (1.36-fold ± 0.09). For comparison, the known Notch activator EDTA upregulated Hes1 (12.00-fold ± 1.30) and Hey1 (6.71-fold ± 0.70) in HUVECs, right graph. *p<0.001. B) PCA plot of RNA-seq comparing EDTA stimulated HUVECs against HBSS stimulated HUVECs after four and six hours. The explained variance ratios are low, and there is no clear clustering of individual groups. This indicates EDTA stimulation does not cause large biological shifts within HUVECs. The graph on the right illustrates the variance explained by the first three principal components. C) qPCR verification of select DEGs from the samples incubated for four hours confirmed statistical significance in the fold change of Jag2 (0.59-fold ± 0.05), Platelet Derived Growth Factor Subunit B (PDGFB) (0.75-fold ± 0.09), Vascular Endothelial Growth Factor Receptor 1 (FLT1) (1.28-fold ± 0.05), Hes4 (2.80-fold ± 0.07), HeyL (12.20-fold ± 0.88), Hey1 (1.75-fold ± 0.03), and Vascular Endothelial Growth Factor A (VEGFA) (1.44-fold ± 0.08). *p<0.05. D) qPCR verification of select DEGs from the samples incubated for six hours confirmed statistical significance in the fold change of Jag2 (0.75-fold ± 0.03), PDGFB (0.71-fold ± 0.07), Hes4 (2.74-fold ± 0.22), and FosB proto-oncogene, AP-1 Transcription Factor Subunit (FosB) (20.08-fold ± 2.98). *p<0.05.
It is important to keep in mind that these genes can also be activated by other pathways when assessing in vitro as well as in vivo studies. We found that, compared to HUVECs without media change, media containing fetal bovine serum (FBS), an ingredient used in EC media, upregulated Hes1 (10.54-fold ± 1.14, p<0.001), Hey1 (14.90-fold ± 1.53, p<0.001), and Hey2 (10.55-fold ± 0.94, p<0.001) (Fig. 3A) one-hour after the media had been changed. To a lesser degree, other ingredients in EC media (vascular endothelial growth factor A (VEGFA), fibroblast growth factor (FGF), insulin-like growth factor 1 (IGF), citric acid, and heparin) increased Hey1 expression (2.20-fold ± 0.14, p<0.001) but did not affect Hes1 or Hey2 expression relative to basal media (Fig. 3A). To account for the effect of flow on Notch activation, media replaced without FBS or growth factors resulted in a modest upregulation of Hey1 (1.36-fold ± 0.09, p<0.001). Woltje et al. reported similar findings and further identified the ALK1 signaling pathway, activated by bone morphogenic proteins in FBS, as the cause for this upregulation. They reported that this effect abates after 3 to 4-hours [80]. Given these findings, culture conditions (for example, replacing media to stimulate cells) and time of RNA extraction are of paramount importance when assessing changes in transcription of downstream Notch targets by qPCR.
RNA Sequencing.
RNA-seq can be performed when an unbiased approach is desirable, such as when testing novel Notch activators or identifying downstream targets in a subset of cells under specific conditions. Genes of interest should then be individually verified by qPCR. EDTA or EGTA can be used as positive controls, bearing in mind that they should be used in conjunction with GSIs to conclusively identify genuine Notch targets. In EDTA stimulated HUVECs incubated for either four or six-hours, we found RNA-seq revealed differentially expressed genes (DEGs) which included canonical downstream targets such as Hes4 (2.80-fold ± 0.07, p<0.00005) and HeyL (12.20-fold ± 0.88, p<0.0005), as verified by qPCR (Fig. 4E). Of note, Principal Component Analysis (PCA) of EDTA stimulated HUVECs revealed a modest separation of experimental groups (0.40 for PC1 and 0.20 for PC2), suggesting EDTA activation of Notch may not elicit a global change in gene expression.
In summary, the findings discussed above illustrate, via usage of positive controls and pharmacological inhibitors, a variety of methods that can be used to detect and quantify Notch activation in vitro. These assays can be particularly useful for studying small molecule activation of Notch. We will use CADASIL, caused by Notch3 inactivation, as an example for this approach. Ye et al. demonstrated that N-methylhemeanthidine chloride (NMHC) activates Notch1 [81]. It may be of interest to assess whether NMHC is capable of activating Notch3 within brain vascular smooth muscle cells and pericytes, and thus protect CADASIL patients from stroke. We recommend isolating brain cells from CADASIL mutant mice, such as [82], for stimulation with NMHC and positive control EDTA. Notch activation can then be confirmed in these cells by a variety of the methods discussed above, such as immunoblotting and immunocytochemistry. Alternatively, transfected human embryonic kidney cells, which do not express endogenous Notch proteins, can provide readouts from the transfected plasmids. A wild type or Notch3 CADASIL mutant plasmid co-transfected with luciferase plasmids can be used to study whether NMHC increases Notch3 activity in the CADASIL mutant cells at comparable levels to cells transfected with wild type plasmids. In vivo experiments should then be performed to test whether NMHC can activate Notch3 and reverse the CADASIL phenotype. Immunohistochemistry and ELISA against the activated form of Notch3 can also be performed. Finally, NMHC reversal of CADASIL phenotype can be assessed through quantification of vascular smooth cell coverage of arterioles in brains and retinas of CADASIL mice.
NOTCH ACTIVATION IN VITRO
Image processing.
Processing and quantifying images focusing on vasculature presents multiple challenges, and there are multiple options to achieve this goal. Digital scanners, such as Pannoramic Viewer (3DHistech), provide free software with quantification capabilities such as distance, perimeter, and area measurements. These features are particularly useful for chromogenic stains, as well as bright fluorescent dyes that do not require high magnification, such as TUNEL stains or Isolectin-B4 tagged with Alexafluor. Another advantage is the ability to organize and visualize multiple tissues simultaneously at a range of magnifications, while digitalization preserves the data. Magnification limits vary, however, and might not be suitable for weak fluorescent intensities. For these cases, as well as for thicker sections requiring confocal microscopy, other software may be more useful. An open-source software appropriate for vascular analysis is ImageJ or Fiji (Fiji stands for Fiji is just ImageJ), (NIH), which provides multiple tools and macros generated by multiple investigators, as well as universities. Some useful examples are manuals generated by Integrated Light Microscopy Core Facility at The University of Chicago (https://voices.uchicago.edu/confocal/image-processing/imagej-fiji-help/). An ImageJ macro generated for cross sections of retinas can be accessed here: TUNEL Cell Counter - RETINA Analysis Toolkit (https://imagej.net/plugins/retina-analysis-toolkit). This provides macros for quantitation of digital red green blue (RGB) images on cryosections. This method would be useful for models of, for example, proliferative diabetic retinopathy using adult mice. However, for developing mouse models, whole mount tissues analyzed by confocal microscopy are required to quantify vascular front vs remodeling areas of the vasculature, a detailed resource for analyzing murine vasculature is [101] using Minkowsky functionals run in MATLAB software. Other useful tools for vascular analysis are: [102] (for zebrafish), and [103] (for brain vessels). Always run tests in subsets of stains in order to ensure the image quality is appropriate for the parameters selected and question asked, in order to avoid incorrect interpretations or measurements.
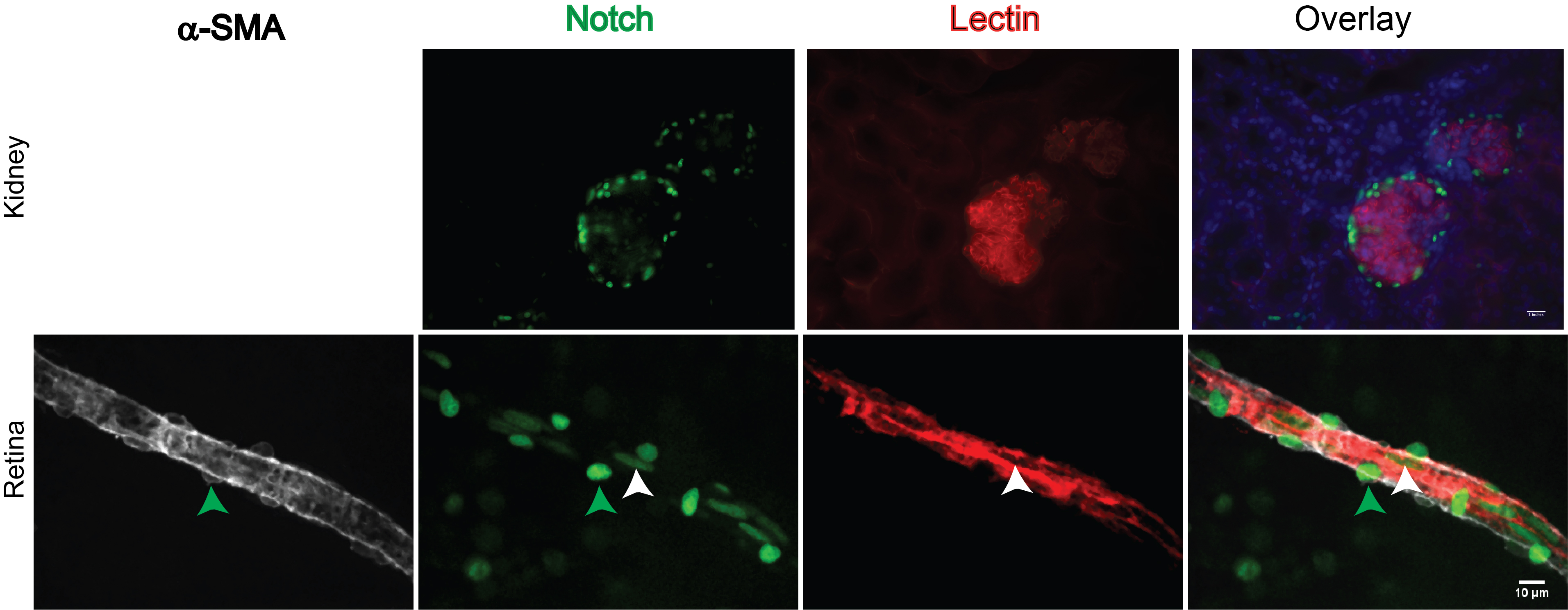
To give an example of how these mouse models could be used: we previously studied whether vascular endothelial growth factor A (VEGF-A) inhibition leads to Notch activation within the tumor vasculature of neuroblastoma xenografts [104]. Tumor bearing mice were treated with the anti-VEGFR2 DC101 for 10 days. IHC against cleaved Notch1 antibody recommended above (Val1744) revealed increased Notch activation within the tumors in response to DC101. Further proof of this activation could be obtained from implanting tumors into a cross of VENUS mice and Rag2 knockout mice. This yields mice with a fluorescent Notch reporter restricted to tumor endothelial cells and murine recruited cells. Notch activation can then be detected by fluorescent microscopy or flow cytometry. Samples should be double labeled with EC markers anti-CD31 or isolectin-B4 prior to visualization or sorting. Treating these mice with DC101 would help visualize and quantify Notch1 activation in tumor ECs as a result of VEGFR2 inhibition. Breeding Ribotag mice crossed with immunodeficient mice would allow RNA extraction from murine ECs to study downstream targets via RNA sequencing. These experiments could help us better understand how Notch participates in escape from anti-angiogenic therapy, such as DC101, in neuroblastoma xenografts.
Conclusion
Much of our understanding of the Notch pathway originates from inhibition and deletion studies. Establishing the appropriate conditions under which to study Notch activation is critical for accurate detection and quantification. Identifying these conditions can be a challenge, and, in this review, we compile relevant background information and summarize assays to aid in this process. Although we focus on ECs, many of these methods can be applied to other cell types, organ systems, developmental stages, or pathologies where Notch is involved.
We propose that the reader consider the following steps prior to evaluating Notch activation within a given system (Figure 6, flow chart). First, identify the appropriate cell type of interest, for example, HUVEC, lung, aortic or other ECs. Begin by characterizing the cell’s Notch expression by PCR, immunoblotting, and/or flow cytometry. Then, establish the appropriate positive and negative controls (fixed ligands, transfection constructs, pharmacologic inhibitors, siRNA, etc.) using relevant in vitro assays, such as immunoblotting, ICC, ELISA, luciferase, qPCR, or RNA seq. Once Notch activation is demonstrated in vitro, move on to an appropriate mouse model and test inhibitors in relevant organ(s) before measuring activation by a compound or under specific conditions. When studying vascular systems, these experiments can then be followed by functional testing of Notch activation in ECs via assays such as fibrin gel bead assay (FIBA), trans migration, tube formation, proliferation (in vitro), or aortic ring assays (ex vivo). In vivo assays, such as post-natal retinal development, arterio-venous shunting, or tumor angiogenesis, can also be used. Together, this series of experiments can provide new insights into the Notch pathway and the downstream effects of its activation and contribute to our general understanding of the role of Notch activation in vascular biology.
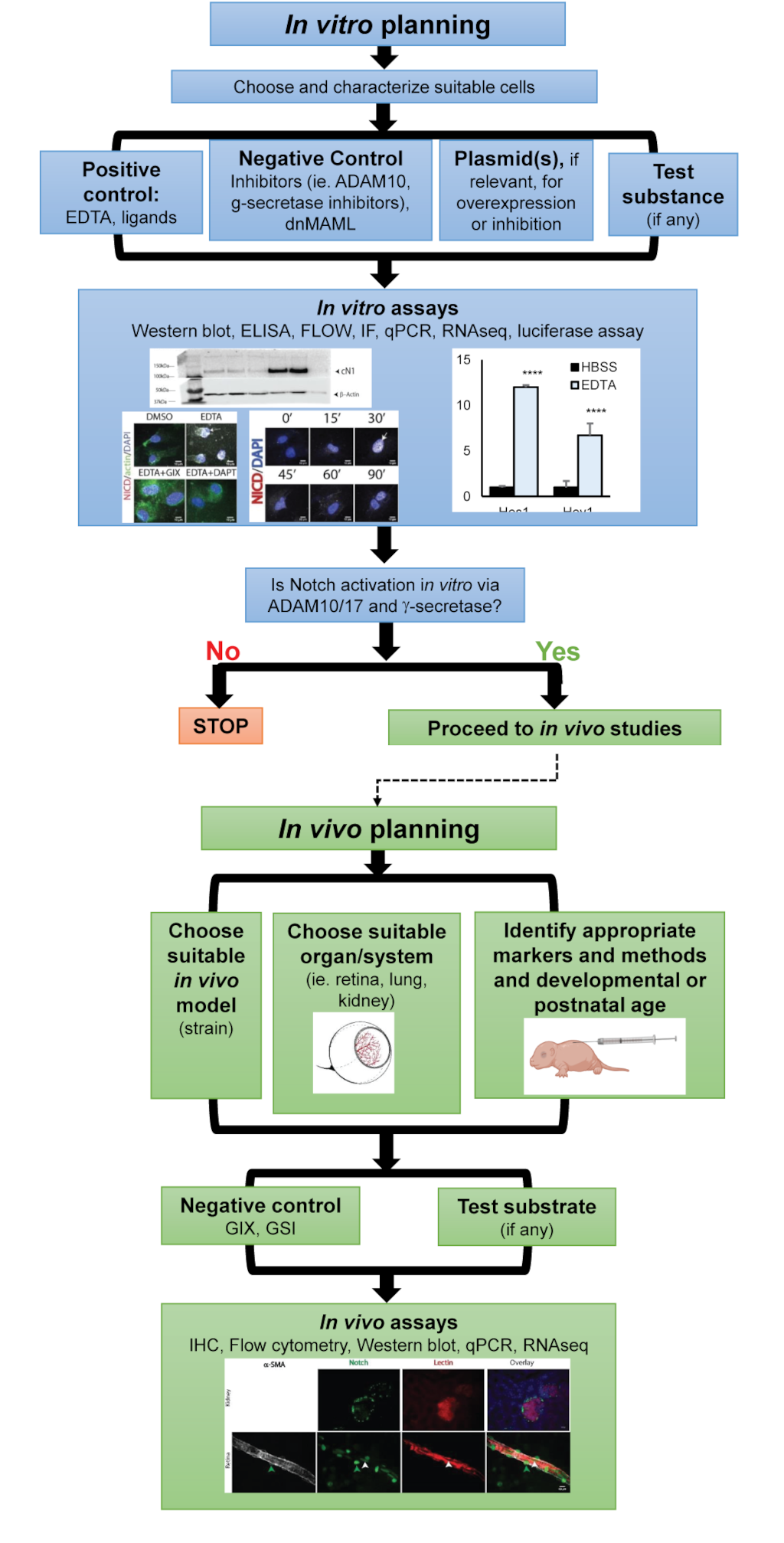
Figure 6: Graphical representation of the proposed sequence of experiments to detect and quantify Notch activation in ECs.
Figure 6: Graphical representation of the proposed sequence of experiments to detect and quantify Notch activation in ECs.
Additional files
Table 3
| Term | Abbreviation | Role within the Notch pathway | Reference |
|---|---|---|---|
| Notch extracellular domain | NECD | Extracellular domain of the Notch receptor | |
| Notch intracellular domain | NICD | Intracellular domain of the Notch receptor | |
| Epidermal growth factor | EGF | Both the extracellular domains of Notch receptors and its ligands contain EGF-like repeats. Notch ligands bind to the NECD via these EGF-like repeats. | |
| Negative regulatory region | NRR | Region between the ligand-binding portion and the transmembrane portion of Notch which prevents exposure of the S2 site until activation occurs |
|
| Lin-12/Notch repeats | LNR | The NRR is made up of three Lin-12/Notch repeats and a heterodimerization domain. The LNR provides structural integrity to the NRR. | |
| Recombination binding protein-Jk-associated molecule | RAM | Domain within the NICD |
|
| Nuclear localization signal | NLS | Directs the NICD to the nucleus after it is released from the cell membrane following g-secretase cleavage |
|
| Proline, glutamic acid, serine, and threonine | PEST | Domain within the NICD |
|
| Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy | CADASIL | Mutations within Notch3 result in CADASIL |
|
| Jagged | Jag | Family of Notch receptors, Jagged 1-2 | |
| Delta-like | Dll | Family of Notch receptors, Delta-like 1 and 3-4 | |
| Delta/Serrate/Lag-2 | DSL | Domain within both families of Notch receptors | |
| A disintegrin and metalloprotease | ADAM | Two members of the ADAM family, ADAM10 and ADAM17/TACE, have been shown to cleave the Notch receptor at site S1 following activation | |
| CBF1, suppressor of hairless, Lag-1 | CSL | CSL is a transcription factor. When Notch is inactive, CSL is bound with co-repressors. Once Notch is activated, the NICD will bind CSL and displace these co-repressors. |
|
| Mastermind/Lag-3 | MAML | Co-activator recruited by the NICD once in the nucleus |
|
| E3 Ligase Suppressor/Enhancer of Lin-12 | SEL-10 | The NICD is eventually ubiquitinated by SEL-10 and degraded. |
|
| Hairy enhancer of split | Hes | Family of basic-helix-loop-helix transcription factors known to be Notch downstream targets | |
| Hes-related with YRPW motif | Hey | Family of basic-helix-loop-helix transcription factors known to be Notch downstream targets |
Table 4
| Term | Abbreviation | Usage pertinent to Notch activation |
|---|---|---|
| Endothelial cell | EC | |
| Human umbilical vein endothelial cell | HUVEC | In vitro model for testing Notch activation within ECs |
| Ethylenediaminetetraacetic acid | EDTA | Known Notch activator which can serve as a useful positive control in vitro |
| Egtazic acid | EGTA | Known Notch activator which can serve as a useful positive control in vitro |
| G1254023X | GIX | ADAM10 and ADAM17 inhibitor |
| g-secretase inhibitors | GSI | g-secretase inhibitors |
| Dibenzazepine | DBZ | g-secretase inhibitor |
| N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester | DAPT | g-secretase inhibitor |
| Cleaved Notch1 | cN1 | Antibodies directed against residues exposed only after S3 cleavage of the N1CD can be used to detect Notch activation |
| Immunocytochemistry | ICC | Technique to visualize antigens in fixed cells |
| 4’,6-diamindino-2-phenylindole | DAPI | Nuclear dye useful when imaging endothelial cells |
| Hank’s buffered saline | HBSS | Negative control |
| Fetal bovine serum | FBS | Common ingredient within cell media that activates Notch |
| Vascular endothelial growth factor A | VEGFA | Common growth factor within endothelial cell media |
| Fibroblast growth factor | FGF | Common growth factor within endothelial cell media |
| Insulin-like growth factor 1 | IGF | Common growth factor within endothelial cell media |
| Notch1 intracellular domain | N1ICD | N1ICD can be overexpressed in mice |
| Locus of X-over P1 | LoxP | Site cleaved by Cre enzyme, used for recombination within the Cre-LoxP system |
| Staphylococcus aureus alpha toxin | Hla | Hla activates Notch in ECs, making it a potential positive control for Notch activation in the postnatal retina model and HUVEC |
Acknowledgements
The Genomics Facility, Center for Research Informatics, Human Tissue Resource Center (HTRC), and Integrated Light Microscopy Core Facility at The University of Chicago.
The authors would like to acknowledge William J. Biermann for his support on the graphic rendering of the retina in Figure 6.
Authors' contributions
Conception and design: SH, LW, JK
Development of methodology: LW, HB, SSH, SH, RK
Acquisition of data: LW, HB, SSH, SH, RK, TG, MN, PH, AMD, NB
Analysis and interpretation of data: LW, HB, SSH, SH, RK, TG
Writing, review, and/or revision of the manuscript: LW, HB, SSH, SH, PH, HC, FFG, JK
Funding
Pediatric Cancer Foundation, Sorkin Fellowship, Feis Fellowship, Cancer Center Support Grant (P30CA014599), and Consejo Nacional de Ciencia y Tecnología (CONACyT) #143671
Original submitted files for images
Below are the links to the authors’ original submitted files for images.
Original image file for Figure 1
Original image file for Figure 2
Original image file for Figure 3
{kind=link}
Original image file for Figure 4
Original image file for Figure 5
{kind=link}
Original image file for Figure 6
References
- Selective repression of Notch pathway target gene transcription. Dev Biol. 2011;360(1):123-31.
- The molecular logic of Notch signaling--a structural and biochemical perspective. J Cell Sci. 2008;121(19):3109-19.
- Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8(6):707-19.
- Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14(11):1343-52.
- Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci U S A. 2001;98(10):5643-8.
- Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270-4.
- Induced pluripotent stem cells with NOTCH1 gene mutation show impaired differentiation into smooth muscle and endothelial cells: Implications for bicuspid aortic valve-related aortopathy. J Thorac Cardiovasc Surg. 2018;156(2):515-522.
- Mutations in NOTCH1 cause Adams-Oliver syndrome. American journal of human genetics. 2014;95(3):275-284.
- Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707-10.
- Notch3 is necessary for blood vessel integrity in the central nervous system. Arterioscler Thromb Vasc Biol. 2015;35(2):409-20.
- Combined deficiency of Notch1 and Notch3 causes pericyte dysfunction, models CADASIL, and results in arteriovenous malformations. Sci Rep. 2015;5:16449-.
- The developmental biology of genetic Notch disorders. Development. 2017;144(10):1743-1763.
- Jagged1 (JAG1): Structure, expression, and disease associations. Gene. 2016;576(1):381-4.
- A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat Struct Mol Biol. 2008;15(8):849-57.
- Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell. 1991;67(4):687-99.
- Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101(45):15949-54.
- Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 2004;18(20):2474-8.
- Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445(7129):776-80.
- Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature. 2006;444(7122):1032-7.
- Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444(7122):1083-7.
- The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137(6):1124-35.
- Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A. 2008;105(6):1955-9.
- JAGGED1 expression in human embryos: correlation with the Alagille syndrome phenotype. J Med Genet. 2000;37(9):658-62.
- Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822-9.
- Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8(5):723-30.
- Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood. 2012;119(9):2149-58.
- Patent ductus arteriosus in mice with smooth muscle-specific Jag1 deletion. Development. 2010;137(24):4191-9.
- NOTCH regulation of the endothelial cell phenotype. Curr Opin Hematol. 2018;25(3):212-218.
- Inhibitory role of Notch1 in calcific aortic valve disease. PloS one. 2011;6(11):e27743-e27743.
- High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005;65(18):8530-7.
- NOTCH pathway activation in infantile hemangiomas. Journal of Vascular Surgery: Venous and Lymphatic Disorders. 2021;9(2):489-496.
- A switch in Notch gene expression parallels stem cell to endothelial transition in infantile hemangioma. Angiogenesis. 2010;13(1):15-23.
- JAGGED1 Signaling Regulates Hemangioma Stem Cell–to–Pericyte/Vascular Smooth Muscle Cell Differentiation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(10):2181-2192.
- The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc Natl Acad Sci U S A. 1998;95(14):8108-12.
- Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell. 1997;90(2):281-91.
- Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development. 2004;131(21):5367-80.
- Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. 2000;127(7):1373-85.
- Epsin-Dependent Ligand Endocytosis Activates Notch by Force. Cell. 2017;171(6):1383-1396 e12.
- Mechanisms of Notch signaling: a simple logic deployed in time and space. Development. 2019.146(3).
- Mechanism and significance of cis-inhibition in Notch signalling. Curr Biol. 2011;21(1):R40-7.
- Cis-activation in the Notch signaling pathway. Elife. 2019.8.
- The "a disintegrin and metalloprotease" (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20(2):126-37.
- ADAM10 and ADAM17 have opposite roles during sprouting angiogenesis. Angiogenesis. 2015;18(1):13-22.
- The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11(21):2615-24.
- Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90(2):271-80.
- Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009;284(45):31018-27.
- A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207-16.
- The disintegrin and metalloproteinase ADAM10 mediates a canonical Notch-dependent regulation of IL-6 through Dll4 in human endothelial cells. Biochem Pharmacol. 2014;91(4):510-21.
- Staphylococcus aureus alpha toxin activates Notch in vascular cells. Angiogenesis. 2019;22(1):197-209.
- Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009;29(21):5679-95.
- Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393(6683):382-6.
- Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 1999;13(21):2801-10.
- Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci U S A. 1999;96(12):6959-63.
- Signalling downstream of activated mammalian Notch. Nature. 1995;377(6547):355-8.
- Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell. 2006;124(5):973-83.
- A novel hes5/hes6 circuitry of negative regulation controls Notch activity during neurogenesis. Dev Biol. 2005;281(2):318-33.
- The Hes gene family: repressors and oscillators that orchestrate embryogenesis. Development. 2007;134(7):1243-51.
- Identification of Notch target genes in uncommitted T-cell progenitors: No direct induction of a T-cell specific gene program. Leukemia. 2006;20(11):1967-77.
- Ligand-receptor interactions and trans-endocytosis of Delta, Serrate and Notch: members of the Notch signalling pathway in Drosophila. J Cell Sci. 1999;112 ( Pt 19):3289-97.
- sel-10, a negative regulator of lin-12 activity in Caenorhabditis elegans, encodes a member of the CDC4 family of proteins. Genes Dev. 1997;11(23):3182-93.
- The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev Cell. 2004;6(2):183-92.
- Soluble Notch ligand and receptor peptides act antagonistically during angiogenesis. Cardiovasc Res. 2015;107(1):153-63.
- Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J Cell Sci. 2000;113 Pt 23:4313-8.
- Calcium depletion dissociates and activates heterodimeric notch receptors. Mol Cell Biol. 2000;20(5):1825-35.
- Nuclear magnetic resonance structure of a prototype Lin12-Notch repeat module from human Notch1. Biochemistry. 2003;42(23):7061-7.
- Evidence for increased exposure of the Notch1 metalloprotease cleavage site upon conversion to an activated conformation. Structure. 2011;19(4):546-54.
- Notch activation stimulates transient and selective binding of Su(H)/CSL to target enhancers. Genes Dev. 2007;21(11):1322-7.
- Notch1 functions as a negative regulator of lymphatic endothelial cell differentiation in the venous endothelium. Development. 2013;140(11):2365-76.
- Notch alters VEGF responsiveness in human and murine endothelial cells by direct regulation of VEGFR-3 expression. J Clin Invest. 2007;117(11):3369-82.
- Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: role of p21Cip1 repression. Mol Cell Biol. 2004;24(20):8813-22.
- Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screen. 2005;8(2):161-71.
- Gamma secretase inhibitors of Notch signaling. Onco Targets Ther. 2013;6:943-55.
- Notch inhibition as a promising new approach to cancer therapy. Adv Exp Med Biol. 2012;727:305-19.
- NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov. 2015;5(2):182-97.
- Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464(7291):1052-7.
- Blood biomarkers in a mouse model of CADASIL. Brain Research. 2016;1644:118-126.
- The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18(8):901-11.
- Notch signaling in primary endothelial cells. Ann N Y Acad Sci. 2003;995:162-70.
- Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun. 2000;275(2):652-60.
- Serum induces transcription of Hey1 and Hey2 genes by Alk1 but not Notch signaling in endothelial cells. PLoS One. 2015;10(3):e0120547-.
- Small molecule activation of NOTCH signaling inhibits acute myeloid leukemia. Scientific Reports. 2016;6(1):26510-.
- Transgenic Mice Expressing Mutant Notch3 Develop Vascular Alterations Characteristic of Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy. The American Journal of Pathology. 2003;162(1):329-342.
- Groves, Overview of genetic tools and techniques to study Notch signaling in mice. Methods Mol Biol. 2014;1187:47-61.
- Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100(25):14920-5.
- C-terminal deletion of NOTCH1 intracellular domain (N1ICD) increases its stability but does not amplify and recapitulate N1ICD-dependent signalling. Scientific Reports. 2017;7(1):5034-.
- Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res. 2008;103(4):423-31.
- Notch1 activation in embryonic VE-cadherin populations selectively blocks hematopoietic stem cell generation and fetal liver hematopoiesis. Transgenic Res. 2013;22(2):403-10.
- A BAC transgenic Hes1-EGFP reporter reveals novel expression domains in mouse embryos. Gene Expr Patterns. 2011;11(7):415-26.
- Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nature Communications. 2017;8(1):1574-.
- Rapid identification of homologous recombinants and determination of gene copy number with reference/query pyrosequencing (RQPS). Genome Res. 2009;19(11):2081-9.
- Cre-conditional expression of constitutively active Notch1 in transgenic mice. Genesis. 2007;45(5):259-65.
- Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6(3):314-22.
- A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Developmental Biology. 2013;13(1):15-.
- Nas transgenic mouse line allows visualization of Notch pathway activity in vivo. Genesis. 2006;44(6):277-86.
- Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106(33):13939-44.
- Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood. 2008;112(5):1720-9.
- Therapeutic antibody targeting of Notch3 signaling prevents mural cell loss in CADASIL. J Exp Med. 2017;214(8):2271-2282.
- Notch receptor regulation of intestinal stem cell homeostasis and crypt regeneration. Dev Biol. 2015;402(1):98-108.
- Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development. 2012;139(3):488-97.
- The mouse retina in 3D: quantification of vascular growth and remodeling. Integr Biol (Camb). 2013;5(12):1426-38.
- Development of an ImageJ-based method for analysing the developing zebrafish vasculature. Vasc Cell. 2017;9(1):2-.
- A Practical Guide to the Automated Analysis of Vascular Growth, Maturation and Injury in the Brain. Front Neurosci. 2020;14:244-.
- Vascular endothelial growth factor blockade rapidly elicits alternative proangiogenic pathways in neuroblastoma. Int J Oncol. 2009;34(2):401-7.

The present article has been published in Vascular Cell journal by Publiverse Online S.R.L.